 Arthritis Foundation, Alabama Chapter
Arthritis Foundation, Alabama Chapter
Professor of Pediatrics & Medicine
Director, Pediatric Rheumatology
| Address: | 1825 University Blvd Shelby Building, 306 UAB Birmingham, AL 35294 |
| Telephone: | (205) 996-9191 |
| FAX: | (205) 996-9545 |
| Email: | rcron@peds.uab.edu |
| Members of the Laboratory |
| Publications |
__________________________________________________________
Education
B.S. (Biomedical Sciences), University of California, Riverside, CA
Pre-doctoral fellow (Howard Hughes Medical Institute), National Institutes of Health, Bethesda, MD
Ph.D. (Immunology), University of Chicago, Chicago, IL
M.D. UCLA, Los Angeles, CA
Residency (Pediatrics), Stanford Children’s Hospital, Palo Alto, CA
Fellowship (Pediatric Rheumatology), University of Washington, Seattle, WA
Post-doctoral Fellow (Howard Hughes Medical Institute scholar), Stanford University, Palo Alto, CA
Research Interests
Host transcription factors exploited by HIV-1. HIV-1, the cause of AIDS, has infected over 40 million individuals world-wide. Although vast improvements in therapy have been developed over the last decade, HIV-1 cannot be totally eliminated from the host due to its ability to enter a resting or latent state in 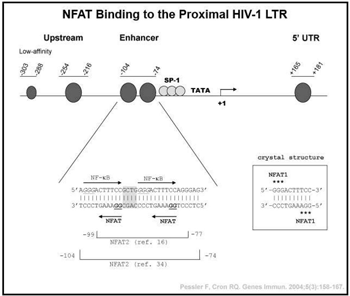 CD4 T cells. Because HIV-1 relies on host transcription factors to replicate, we are exploring the role of the calcium activated nuclear factor of activated T cells (NFAT) transcription factors in regulating HIV-1 transcription. We and others have shown that the CsA-sensitive NFAT proteins bind to the proximal HIV-1 promoter/long terminal repeat (LTR) in vitro and up-regulate HIV-1 transcription. We have further demonstrated that NFAT proteins bind to the integrated HIV-1 LTR in primary human CD4 T cells in vivo by chromatin immunoprecipitation, and this binding is disrupted by the regulatory T cell transcription factor, FOXP3. In addition, we are attempting to exploit NFAT activation as a means of activating HIV-1 LTR activity in latently infected cells. Recently, we identified a novel binding site for the c-maf transcription factor located adjacent to the proximal NFAT sites in the HIV-1 LTR. Our studies reveal synergistic transcriptional activation and increased infection of HIV-1 by c-maf, NFAT2, and NFΚB p65 in primary human IL-4-producing CD4 T cells. Thus, c-maf will likely be a novel therapeutic target in the treatment of HIV-1.
CD4 T cells. Because HIV-1 relies on host transcription factors to replicate, we are exploring the role of the calcium activated nuclear factor of activated T cells (NFAT) transcription factors in regulating HIV-1 transcription. We and others have shown that the CsA-sensitive NFAT proteins bind to the proximal HIV-1 promoter/long terminal repeat (LTR) in vitro and up-regulate HIV-1 transcription. We have further demonstrated that NFAT proteins bind to the integrated HIV-1 LTR in primary human CD4 T cells in vivo by chromatin immunoprecipitation, and this binding is disrupted by the regulatory T cell transcription factor, FOXP3. In addition, we are attempting to exploit NFAT activation as a means of activating HIV-1 LTR activity in latently infected cells. Recently, we identified a novel binding site for the c-maf transcription factor located adjacent to the proximal NFAT sites in the HIV-1 LTR. Our studies reveal synergistic transcriptional activation and increased infection of HIV-1 by c-maf, NFAT2, and NFΚB p65 in primary human IL-4-producing CD4 T cells. Thus, c-maf will likely be a novel therapeutic target in the treatment of HIV-1.
Genetic defects in lymphocyte cytolysis in macrophage activation syndrome. Macrophage activation syndrome (MAS) is a hyper-inflammatory immune response in children and adults that is often triggered by certain infectious (e.g. EBV), autoimmune (e.g. lupus), autoinflammatory (e.g. Still disease), and oncologic (e.g. T cell leukemia) disorders. MAS results in pro-inflammatory cytokine storm leading to pancytopenia, coagulopathy, central nervous system dysfunction, and multi-organ system failure. MAS is frequently lethal like its cousin disease familial hemophagocytic lymphohistiocytosis (fHLH). fHLH is uniformly fatal if not treated aggressively and typically presents in the first few months of life in infants 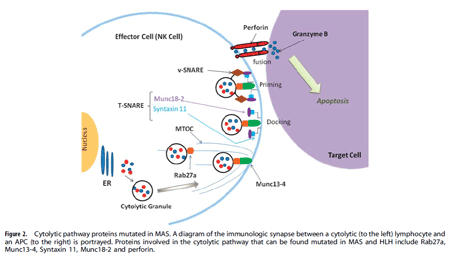 with bi-allelic genetic defects in one of the proteins involved in perforin mediated cytolysis by natural killer (NK) cells and CD8 cytotoxic lymphocytes. Recently, mono-allelic (heterozygous) mutations in cytolytic pathway proteins (e.g. perforin, Munc13-4, Rab27a, etc.) have been identified in a substantial percentage of MAS patients presenting beyond infancy. In our MAS patient cohort, we have identified several mutations, including novel mutants, in a variety of cytolytic pathway genes. Using lentiviral transduction of mutant and wild-type genes into NK cells, we demonstrate decreased cytolytic activity by over-expression of the mutant genes, suggesting a partial dominant-negative effect. These studies suggest that there are likely genetic predispositions to develop MAS, and we are currently exploring the novel mutations and their pathophysiological consequences on lymphocyte mediated cytolytic function.
with bi-allelic genetic defects in one of the proteins involved in perforin mediated cytolysis by natural killer (NK) cells and CD8 cytotoxic lymphocytes. Recently, mono-allelic (heterozygous) mutations in cytolytic pathway proteins (e.g. perforin, Munc13-4, Rab27a, etc.) have been identified in a substantial percentage of MAS patients presenting beyond infancy. In our MAS patient cohort, we have identified several mutations, including novel mutants, in a variety of cytolytic pathway genes. Using lentiviral transduction of mutant and wild-type genes into NK cells, we demonstrate decreased cytolytic activity by over-expression of the mutant genes, suggesting a partial dominant-negative effect. These studies suggest that there are likely genetic predispositions to develop MAS, and we are currently exploring the novel mutations and their pathophysiological consequences on lymphocyte mediated cytolytic function.
Personal Interests
My family, travel, skiing (water and snow), rock and roll music, golf.