
 Although unproven, this novel sickle cell therapy serves as a potential cure. More measures need to be taken to determine long-term function and organ improvement.New research from University of Alabama at Birmingham, published in the New England Journal of Medicine, suggests a gene therapy called LentiGlobin could provide a permanent cure for sickle cell disease.
Although unproven, this novel sickle cell therapy serves as a potential cure. More measures need to be taken to determine long-term function and organ improvement.New research from University of Alabama at Birmingham, published in the New England Journal of Medicine, suggests a gene therapy called LentiGlobin could provide a permanent cure for sickle cell disease.
Julie Kanter, M.D., director of the UAB Adult Sickle Cell Clinic, says patients treated with this therapy are beginning to show signs of producing stable amounts of normal red blood cells containing hemoglobin.
SCD occurs in about one out of every 365 Black or African American births, according to the Centers for Disease Control and Prevention, and about one in 13 Black or African American babies is born with sickle cell trait.
How the therapy works
Kanter says there are several types of gene therapy (gene addition/transfer, gene editing, gene correction and gene silencing), but this particular therapy is gene addition or transfer.
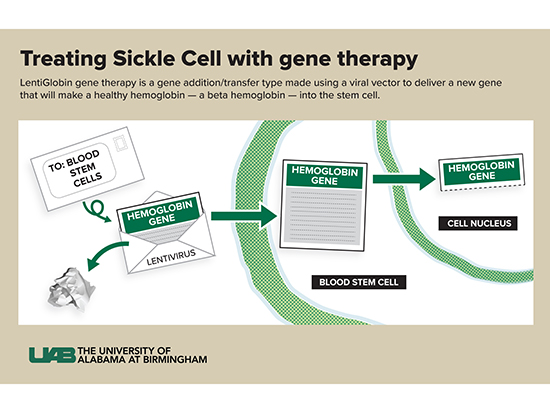
“In this therapy, we do not change or edit the gene that causes sickle cell disease,” Kanter said. “Instead, we use a viral vector to deliver a new gene that will make a healthy hemoglobin — a beta hemoglobin — into the stem cell. This is like coding new instructions into the cell. The old instructions for hemoglobin S are still there, but now the cell can make HbA and HbS. The vector can deliver more than one copy of the instructions to each cell — usually between one and four copies — so the cell can make more HbA than HbS.
 What is a vector?
What is a vector?
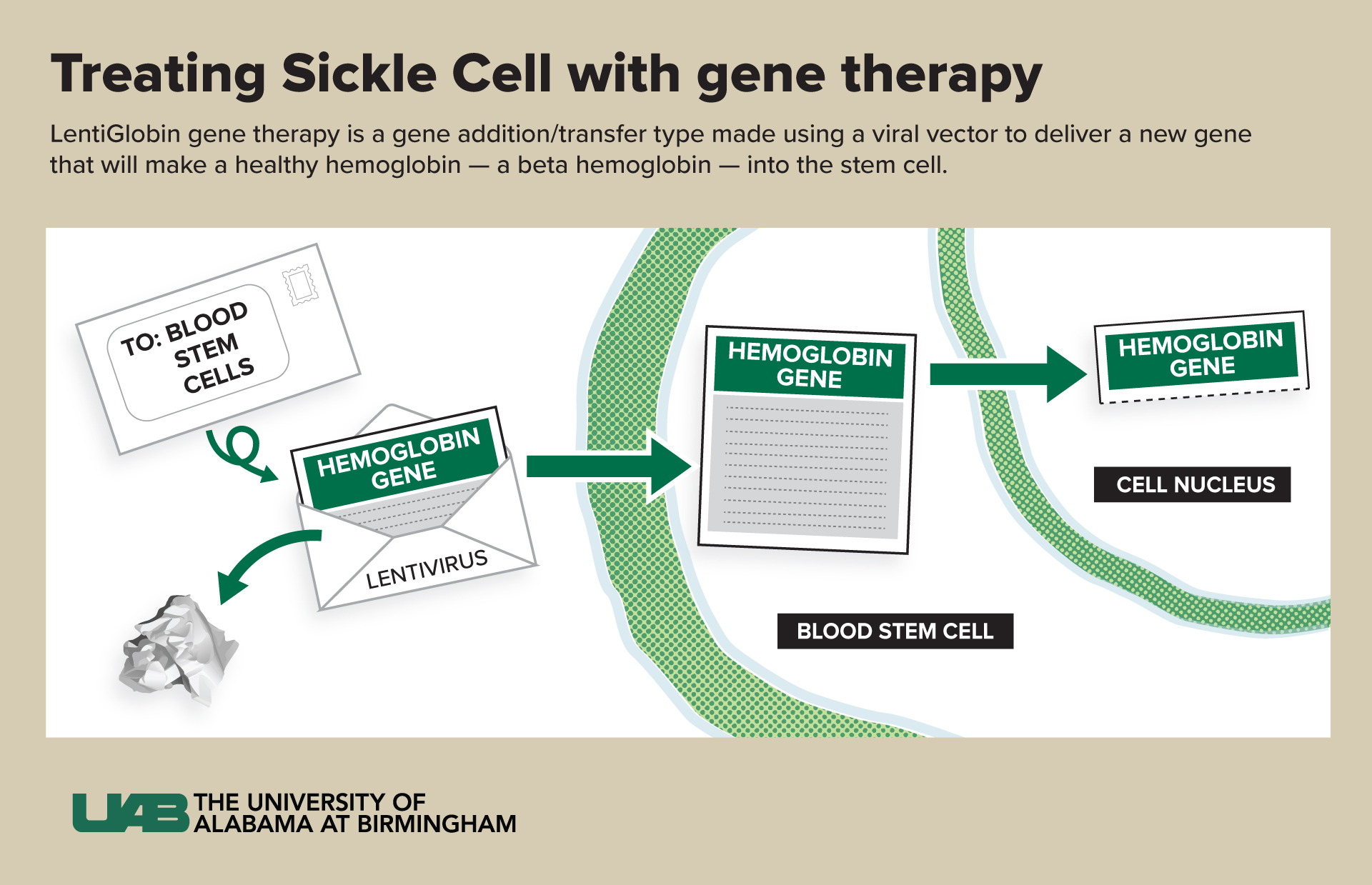
A vector is part of a virus. Kanter compares vectors to envelopes and letters.
“I like to think of it as an envelope,” she said. “We take out the bad part of a virus (the letter) and leave the empty envelope. We put a new gene (the new letter) with the right instructions into the envelope and send it into the stem cells. The viral parts of the letter are removed so patients don’t get the virus itself — they only get the letter coding for the new hemoglobin, called HbAT87Q.”
What is T87Q?
T87Q is a special type of hemoglobin A that is slightly different from regular hemoglobin A and has two advantages:
- The intentional change inserted (called T87Q) makes the hemoglobin even less likely to cause sickling when it is near a hemoglobin S.
- The HbAT87Q can also be measured more accurately inside the cell (since it is slightly different from regular hbA), which allows doctors to know how much of the new hemoglobin a patient is making compared to how much they get from a transfusion.
 Julie Kanter, M.D.
Julie Kanter, M.D.
(Photography: Steve Wood)Next steps
Kanter says that, although this therapy is providing a significant amount of hope, researchers continue to test to make sure the therapy remains safe.
“In an earlier part of this study, we were not able to get enough of the new gene into each cell,” Kanter said. “Not enough envelopes were delivered.”
This caused the stem cells to be extra stressed and the patients to still have some parts of sickle cell disease. They had only slight improvements compared to group C.
“Unfortunately, the stressed-out cells are also more likely to make bad clones, which can cause cancer,” she said. “Two patients in group A developed leukemia because the cells were too stressed.”
It is important to note that this was not caused by the viral vector or the new gene (not from the LentiGlobin) but from the stress of the procedure and the insufficient cell correction.
“We need to see that we have fixed this problem in group C — and no one else develops leukemia,” Kanter said. “We also need to make sure this procedure both reduces pain/stops all pain crisis and prevents organ damage from sickle cell. This will take time. We will have to watch people for the next two to 15 years and measure their organ function compared to those who did not get this therapy.”
Hope for the future
Much of Kanter’s career has been dedicated to helping those with SCD. A therapy like this is a game changer, according to Kanter.
“It means a lot,” she said. “People with sickle cell disease have endured unnecessary hardship for more than 100 years. They have fewer medications and therapies than many other diseases and have received much less attention and funding. We need new and better options for people with sickle cell disease.”
She also says this is just a beginning.
“This therapy gives us hope and promise for a better future for those living with sickle cell disease.”
“We need to make these treatments available, and we need all people with sickle cell disease to have a sickle cell doctor to make that happen,” she said. “We need the therapy to be affordable so that people everywhere living with this disease have the option for gene therapy. Right now, most people with sickle cell disease live in sub-Saharan Africa and in India. They don’t have even the basic treatments they need like vaccines, penicillin or hydroxyurea that can make a huge difference in people’s lives with SCD. Eventually we need people in these areas to have equal opportunity to better outcomes.”