
Researchers at the University of Alabama at Birmingham (UAB) may have found a new approach to countering cholesterol-related disease in blood vessels.
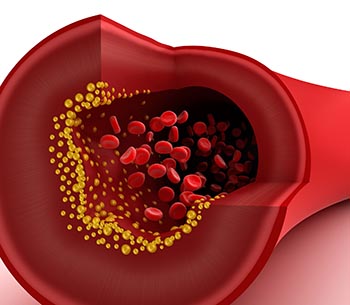
Chemical changes to a key gene were found to encourage the fatty buildup in blood vessels behind atherosclerosis, a major cause of heart attack and stroke. The researchers are excited because the newfound, disease-causing mechanism is reversible – and by dietary changes as much as drug treatments.
The American Heart Association (AHA) shared their excitement enough to ask the team to present their data during the AHA’s recent Emerging Science Series event in Dallas, a presentation that was also broadcast as a webinar.
Many environmental factors, including diet, are known to influence blood cholesterol levels, but much of the genetic contribution to each person’s lipid profile remains unexplained.
“New technologies and study models are expanding our knowledge about the genetic architecture of lipids,” said lead study author Stella Aslibekyan, Ph.D., assistant professor in the Department of Epidemiology within the UAB School of Public Health. “These lessons promise to reveal new targets for drug design and to inform future efforts to prevent cardiovascular disease.”
Important blind spot
Going into the current study, an early clue to genetic influence over blood lipids was related to the enzyme carnitine palmitoyltransferase 1A (CPT1A). Changes in the gene coding for this enzyme had been shown to cause higher blood levels of triglycerides, a type of lipid that contributes to cardiovascular disease. CPT1A regulates how cells burn fats for energy.
Genes like CPT1A are DNA chains that hold the instructions for the building of proteins, the workhorse molecules that make up the body's structures and carry its messages. To pass on instructions, genes must be copied, but the process is not perfect. Random changes called mutations are constantly introduced, some of which confer advantages and others cause disease. Mutations in CPT1A, for instance, create a higher risk for metabolic disease in people of Inuit ancestry.
|
“New technologies and study models are expanding our knowledge about the genetic architecture of lipids,” said lead study author Stella Aslibekyan, Ph.D., assistant professor in the UAB School of Public Health. “These lessons promise to reveal new targets for drug design and to inform future efforts to prevent cardiovascular disease.” |
The current study found that a mechanism independent of previously identified mutations is also driving cardiovascular disease through its impact on CPT1A. The results reflect the discovery that, along with random copying mistakes in genes, another more subtle set of mechanisms is affecting gene performance.
So-called epigenetic changes are chemical reactions that turn genes on and off without changing the DNA code we inherit from our parents. Among the most important of these is methylation, the attachment of a methyl group (one carbon and three hydrogens) to certain spots on the DNA chain. The process can turn surrounding genes off, while de-methylation can turn them back on. This back and forth gives genes a way to respond quickly to a changing environment.
The current study determined whether or not more than 470,000 known sites were methylated in DNA of patients taking part in the Genetics of Lipid Lowering Drugs and Diet Network Study (GOLDN Study).
The team then fed the GOLDN data from 888 patients into statistical analyses looking for associations between methylation at any of these sites, as well as higher or lower levels of cholesterol subtypes and triglycerides in a patient’s blood. They found that methylation at just two of 470,000 markers tested – cg00574958 and cg17058475 – within the CPT1A gene were strongly associated with higher levels of triglycerides and very low density lipoproteins (VLDL), known contributors to atherosclerotic disease.
In the case where the CPT1A gene is methylated and shut down, not enough of the CPT1A enzyme is produced. Without the enzyme, carnitine cannot attach to dietary fatty acids, and they cannot be converted into energy without the carnitine tag. Fatty acids then build up, in turn raising levels of triglycerides and VLDL. The study results may inform the search for a demethylase that counters the effect of methylated CPT1A on triglyceride levels.
“Evidence is mounting that the foods we choose to eat constantly change the performance of our genetic material, and in ways that drive risk for diseases from cancer to heart attack,” said Donna Arnett, Ph.D., chair of the UAB Department of Epidemiology, study author and outgoing president of the American Heart Association. Both GOLDN and the current study were led out of her lab. “Epigenetic science has had a huge impact on developmental biology, neurology and cancer research, but is just now gaining speed in cardiology, where its effect on cholesterol levels has been a blind spot.”
Also making an important contribution to the work was Ryan Irvin, Ph.D., assistant professor in UAB Department of Epidemiology.