
 Shanrun Liu, John Mountz and Hui-Chen Hsu in the UAB Comprehensive Flow Cytometry Core, which houses a core facility for single-cell sequencing. The science made possible by sequencing individual cells "is just amazing," Mountz said. "You can really tell what genes are being expressed — not what the potential functional elements are at the DNA level, but what is actually steering the cell.” There are 37.2 trillion cells in the human body, according to a 2013 study from Italy’s University of Bologna. If each cell was a slice of thick-cut bologna, the pile would stretch more than halfway to the sun.
Shanrun Liu, John Mountz and Hui-Chen Hsu in the UAB Comprehensive Flow Cytometry Core, which houses a core facility for single-cell sequencing. The science made possible by sequencing individual cells "is just amazing," Mountz said. "You can really tell what genes are being expressed — not what the potential functional elements are at the DNA level, but what is actually steering the cell.” There are 37.2 trillion cells in the human body, according to a 2013 study from Italy’s University of Bologna. If each cell was a slice of thick-cut bologna, the pile would stretch more than halfway to the sun.
Much like processed meats, gene-sequencing can be a questionable mixture. Every one of your cells, in its nucleus, has a copy of your entire genome, all 3 billion or so A-C-T-G base pairs. But every cell is different. For starters, there are roughly 200 different types of cells — blood cells, nerve cells, muscle cells, skin cells, stem cells, fat cells and so on. And even within the same cell type, lots of different things are going on — that is, the 20,000 genes in your genome are turned on and off in different combinations. It’s the way the body can make everything from a neuron to an egg cell using the same genome.
From smoothie to fruit salad
But traditional gene-sequencing obliterates much of this variety. A saliva sample or cheek swab may contain millions of cells. In order to get at their DNA, labs induce all of those cells to burst open, then they sequence all the genetic code that spills out. This works out fine if you are interested in the genome itself. But if you’re curious about which genes are actually switched on in those cells, you will want to sequence the RNA transcripts that are present. (RNA, you’ll recall, transcribes the DNA code and shuttles it out of the nucleus where the code is used to manufacture the proteins that actually do the work of the cell.) Researchers compare it to a smoothie, with RNA transcripts from a host of individual cells all blended together in a mush.
Single-cell sequencing, on the other hand, is like a fruit salad. Each individual cell in a sample can be handled separately. And when you do it this way, surprising things happen. “Single-cell sequencing gives us a much better window to understand how cells function in their microenvironment,” said Kent Keyser, Ph.D., assistant vice president for research at UAB. “And it is revealing new types of cells — cells that nobody had any idea existed that actually play key roles. This is an incredibly exciting time.”
‘Amazing’ science
| “There’s a whole bunch of low-hanging fruit in every area of science. Today, practically anything you do will be a discovery — it hasn’t been done before at this level. In five years you’ll be repeating what other people have done, but right now we’re at a moment that is wide open.” |
“Single-cell sequencing is revolutionizing immunology,” said John Mountz, M.D., Ph.D., Goodwin-Blackburn Chair and Professor in the Department of Medicine Division of Clinical Immunology and Rheumatology. “The science is just amazing. You can really tell what genes are being expressed — not what the potential functional elements are at the DNA level, but what is actually steering the cell. Immunology is not being done the way it was 10, five, even three years ago.” (For the most part, the single-cell sequencing we’re talking about here is RNA-sequencing, or single-cell RNAseq, but there are other varieties, including V(D)J sequencing to profile the adaptive immune repertoire and ATAC-seq — Assay for Transposase-Accessible Chromatin using sequencing — to understand chromatin accessibility.)
In some ways, that’s just more of the same for Mountz. “Every decade or so there’s another revolution in immunology,” he said. “Just during my career, I’ve seen this go from northern blot to real-time PCR looking at smaller amounts of RNA, to flow cytometry looking at 50 markers per cell, to single-cell, where we can have a complete characterization of an individual cell. We think that in five years you will not be able to produce a first-rate paper or get a top grant without this technology.”
Five years ago, Mountz’s lab dove into single-cell work, “bootstrapping with our own money and equipment that we had to write a grant to obtain,” he said. “We found that we can take a purified population of B cells, as pure as we can make it, and nevertheless every cell is different from every other cell. You can subset that into five to 10 subpopulations, each of which has the potential to either expand or go away. This is a more informative application of what’s going on by a factor of 10.”
Mountz’s lab is studying “the population of cells in patients who have systemic lupus erythematosus compared to cells from people who don’t have SLE,” he said. “You can do that if you can see each cell individually. And once you can do that, you can see what are the pathogenic properties that drive disease. Is a certain receptor up? Is a transcription factor down? You can look closely at the abnormal population in a big group of cells.” In October 2018, Mountz and colleagues published a paper in the Journal of Immunology reporting distinct B cell subpopulations in SLE patients that were associated with disease severity.
 Researchers across UAB are taking advantage of the single-cell sequencing capabilities on campus. As of this summer, the core has had about 140 users and run some 200 single-cell experiments.
Researchers across UAB are taking advantage of the single-cell sequencing capabilities on campus. As of this summer, the core has had about 140 users and run some 200 single-cell experiments.
‘A biological time machine’
The almost magical thing about single-cell sequencing is that it lets researchers delve into cause-and-effect inside the body like never before. “It’s like a biological time machine,” Mountz said. “You can go forward and backward in time.”
That’s thanks to a software tool called Pseudotime, part of the Monocle package developed by the Trapnell Lab at the University of Washington. Pseudotime uses a machine-learning algorithm to understand the sequence of gene-expression changes present in a biological sample.
“Any kind of cell I’m interested in, at whatever stage, its precursors are also in the body as well,” Mountz explained. Because individual cells are engaged in these changes at different rates, a given sample will include examples of cells at various steps along that path. Pseudotime can sort each cell according to its point on this trajectory. Each cell represents a snapshot of one moment in the process.
By stitching these snapshots together, you can create what is in essence a mini movie, revealing how the fates of different cell types shift across “the spectrum of time and space,” Mountz said. “Everybody knows what the problem cells are, but where did they come from? And what will they do next? We can now start to answer those questions.”
 Liu, the manager and primary consultant for single-cell sequencing in the core, at work on a sample.
Liu, the manager and primary consultant for single-cell sequencing in the core, at work on a sample.
In the beginning
This past year, Mountz started a single-cell sequencing core facility within the Comprehensive Flow Cytometry Core he directs. As of this summer, the core has had about 140 users and some 200 experiments, Mountz said.
Shanrun Liu, M.D., Ph.D., is the manager and primary consultant for single-cell sequencing in the core. Immunologists such as Mountz and Casey Weaver, M.D., Wyatt and Susan Haskell Professor of Medical Excellence in Pathology in the multidisciplinary Program in Immunology, were early adopters. “But now we have worked with researchers studying cancer, the kidneys, lung, pancreas, blood, bone marrow, intestines, skin, eyes, teeth and the brain,” Liu said. The technology is so new that “most people still don’t quite understand about single-cell analysis, but they have very good scientific questions to ask,” Liu said. “Our goal is to help them answer those questions and educate them on the basic principles.”
Getting started in single-cell sequencing can be intimidating. The reagents involved cost thousands of dollars, even if you have access to a machine. The CFCC has three: a workhorse Chromium from 10X Genomics; the Fluidigm C1, which has low throughput but provides full-length mRNA sequencing; and a BD Rhapsody. (See “Which one is right for me?”)
The Chromium uses the latest-generation droplet method, in which cells are combined with molecular barcodes inside individual oil droplets. The cells are burst open within the droplets, tagging all of their constituent parts with unique barcodes. Then a batch of cells can be sequenced and analyzed, using the barcodes to determine which genetic elements were associated with which cell. (See “How does it work?”)
That summary glosses over a host of finicky steps. “You have to have a very good quality of cells in your sample,” Liu said. Fibrous clumps can clog the minuscule channels in a single-cell machine. “And dead cells can generate noisy signals and screw up the whole experiment,” he explained. Producing a sample sometimes can require months of work on the part of an investigator, but Liu is able to provide many helpful tips.
With the sample in hand, Liu begins a preparation process that can involve lots of steps on its own. “It’s amazing to watch,” Mountz said. “Shanrun is one of those gifted people with a rare talent for the type of hand-eye coordination you need for molecular biology. Even though he has done this so many times before, he will go down that list of procedures, ticking them off — line by line. An investigator may have spent months getting that sample ready. He’ll emerge two hours later drenched in sweat.”
Liu’s obsessive attention to detail pays off. “His failure rate is only 1 percent,” Mountz said with amazement. “I know how to do it, but I don’t want to do it.” That’s an attitude shared by many investigators. “If I make one pipetting error, that can scrap the whole thing,” Liu said. “It’s one reason most users don’t want to do this by themselves, so as not to take the risk.”
Building a roadmap
 Single-cell sequencing is "wonderful technology," said Robert Welner, who used it to map bone marrow niche populations in a paper published this summer in Cell Reports.Robert Welner, Ph.D., assistant professor in the Division of Hematology and Oncology and an associate scientist in the O’Neal Comprehensive Cancer Center, brought an inDrop single-cell sequencing machine with him when he came to UAB from Harvard Medical School. In a paper published this year, Welner and his collaborators at Harvard and Beth Israel Deaconess Medical Center used inDrop to map bone marrow niche populations. They were interested in the distinct populations that differentiate from mesenchymal stem cells — bone marrow cells that are not involved in producing red blood cells or immune cells. Mesenchymal stem cells differentiate into cell types that eventually produce bone, fat and cartilage. Dysregulation in this process is linked with cancer, obesity, osteoporosis, aging and tooth loss. (Mapping Distinct Bone Marrow Niche Populations and Their Differentiation Paths was published in the journal Cell Reports in July 2019.)
Single-cell sequencing is "wonderful technology," said Robert Welner, who used it to map bone marrow niche populations in a paper published this summer in Cell Reports.Robert Welner, Ph.D., assistant professor in the Division of Hematology and Oncology and an associate scientist in the O’Neal Comprehensive Cancer Center, brought an inDrop single-cell sequencing machine with him when he came to UAB from Harvard Medical School. In a paper published this year, Welner and his collaborators at Harvard and Beth Israel Deaconess Medical Center used inDrop to map bone marrow niche populations. They were interested in the distinct populations that differentiate from mesenchymal stem cells — bone marrow cells that are not involved in producing red blood cells or immune cells. Mesenchymal stem cells differentiate into cell types that eventually produce bone, fat and cartilage. Dysregulation in this process is linked with cancer, obesity, osteoporosis, aging and tooth loss. (Mapping Distinct Bone Marrow Niche Populations and Their Differentiation Paths was published in the journal Cell Reports in July 2019.)
 The inDrop device in Welner's lab uses an open-source design where "everything is external — you're controlling each one of the pumps," he said. (Gif adapted from video provided by Robert Welner.)“We have lots of questions about inflammation and disease related to bone marrow,” Welner said, “but we didn’t have a roadmap as to what ’normal’ looks like — what are the cells that make up the bone marrow that are not blood cells? Now that we have that roadmap, we can ask, during disease or inflammation how is that altered?”
The inDrop device in Welner's lab uses an open-source design where "everything is external — you're controlling each one of the pumps," he said. (Gif adapted from video provided by Robert Welner.)“We have lots of questions about inflammation and disease related to bone marrow,” Welner said, “but we didn’t have a roadmap as to what ’normal’ looks like — what are the cells that make up the bone marrow that are not blood cells? Now that we have that roadmap, we can ask, during disease or inflammation how is that altered?”
Although both the 10X Chromium and the inDrop are droplet-based, the latter uses an open-source design that means costs are lower, Welner said. Unlike the Chromium, which is “plug and play” for easier use, “here everything is external — you’re controlling each one of the pumps,” he said. This results in far lower usage of costly reagents. His lab has worked with Ron Banerjee, M.D., Ph.D., an assistant professor in the Division of Endocrinology on studies of islet cells in diabetes; Maria Grant, M.D., professor in the Department of Ophthalmology; and Etty (Tika) Benveniste, Ph.D., professor in the Department of Cell, Developmental and Integrative Biology, among others. “We’ll teach you how to use the machine, and by the third time you do it you’re independent,” Welner said.
Single-cell sequencing is “wonderful technology,” he noted, “but the scariest thing from my perspective is you can waste tens of thousands of dollars if you don’t really have a research question. You can run this technology on anything — but that doesn’t mean you should.”
As more single-cell papers are published, it is becoming easier to do initial research that can justify the investment, Welner added. “In the case of Tika’s group, there was a paper in Science in 2019 that, while it was not doing what they wanted to do, included datasets that allowed us to download a subset and say, ‘It looks like you could ask some interesting scientific questions here,’” Welner said.
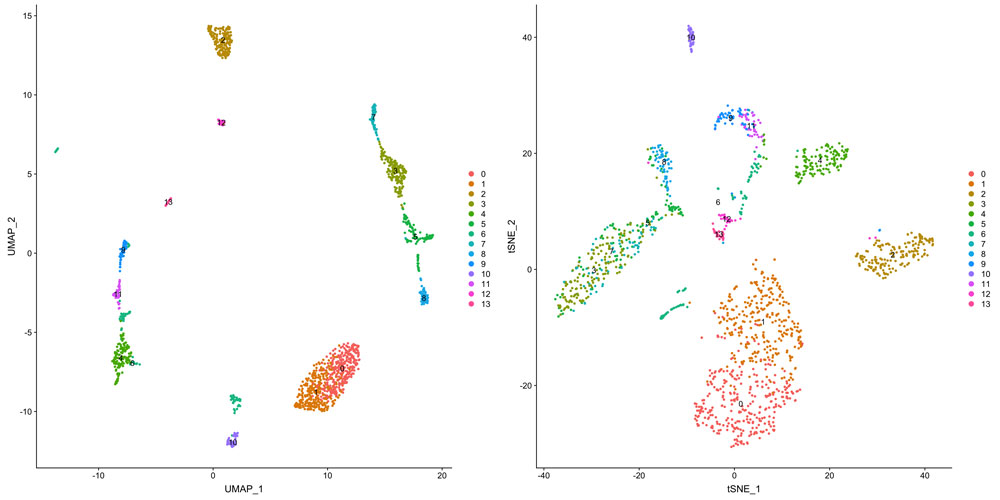 Informaticians such as David Crossman and Min Gao can transform the millions of rows of single-cell sequencing data into graphs like these maps of cell clusters. Image courtesy David Crossman.
Informaticians such as David Crossman and Min Gao can transform the millions of rows of single-cell sequencing data into graphs like these maps of cell clusters. Image courtesy David Crossman.
Analysis dilemmas
Learning to use single-cell machines is one thing, but before researchers can get answers, they have to be able to make sense of the flood of data that comes back.
Another early single-cell paper out of UAB is Single-Cell RNA Sequencing Identifies Candidate Renal Resident Macrophage Gene Expression Signatures across Species, by lead author Bradley Yoder, Ph.D., professor and chair of the Department of Cell, Developmental and Integrative Biology, published by the Journal of the American Society of Nephrology in May 2019. The investigators used single-cell RNA sequencing to identify a novel set of possible cell surface markers to identify macrophage populations in rat, pig and human kidney tissue. They obtained sequencing data from nearly 15,000 individual cells, with means of 54,000 to 112,000 reads per cell, depending on the population.
“There are millions of rows of data,” said Hui-Chen Hsu, Ph.D., an associate professor in the Division of Clinical Immunology and Rheumatology who worked together with Mountz and Liu to bring single-cell sequencing to UAB. “How are you even going to interrogate that? That’s why investigators have to work closely with informaticians.”
In the case of Yoder’s paper that was David Crossman, Ph.D., bioinformatics director at UAB’s Genomic Core and an associate professor in the Department of Genetics. Even for data gurus like Crossman, single-cell sequencing “is a whole different can of worms,” he said. “The data is so cumbersome and so massive. People have developed all kinds of different strategies to analyze it, but it’s the Wild West right now. It reminds me of when bulk RNA-sequencing first came out many years ago, with a new analysis pipeline each week being developed and nobody really knew the best way to do it.”
Crossman has been fielding an increasing number of requests to analyze single-cell data. He is developing a plan for a monthly hackathon-style group that will bring together members of labs across UAB who are involved in or interested in single-cell analysis. “I can teach them how to take the data I crunch and how they can further crunch that,” he said. “If someone in each lab could understand the basics, I could help them with more complex analysis.”
Min Gao, Ph.D., an assistant professor with the UAB Informatics Institute in the School of Medicine, is another key player in the university’s single-cell scene. “I have been working on single-cell sequencing data analysis and the development of single-cell sequencing technologies for several years,” she said. Gao and Mountz recently received an R01 award from the NIH to investigate and compare the factors that are over- and under-expressed in B-cell developmental checkpoints in patients with lupus and healthy donors. “The most rewarding aspect for me is that I can bridge seamlessly between cutting-edge biological and medical research and the most advanced sequencing technologies to help address important scientific questions,” she said.
Gao is a member of the Partnered Advancement of Informatics Research and Support (PAIRS) team at the Informatics Institute, led by Jake Chen, Ph.D., the institute’s associate director and chief bioinformatics officer. “After connecting with research groups, we discuss with the key researchers who generated the data, and we try to learn more information about the samples and the hypothesis for each project,” Gao explained.
Like Welner, Gao is quick to note that “sequencing data is not a panacea,” she said. “It will not address everything.” After she meets with potential collaborators and learns about their expectations, “I will analyze the data and try my best to address all the scientific questions,” she said.
 This cell analysis machine in the Comprehensive Flow Cytometry Core can analyze tens of thousands of cells per second across 50 different dimensions. "Everything we do here is in the thousands," Mountz said.
This cell analysis machine in the Comprehensive Flow Cytometry Core can analyze tens of thousands of cells per second across 50 different dimensions. "Everything we do here is in the thousands," Mountz said.
Data unchained
Analysis is the principal bottleneck to single-cell work. “You cannot only rely on Min Gao or David Crossman to run every analysis,” Hsu said. “And it can take a few months to years to fully appreciate the meaning of the data.” To accelerate that process, Hsu has been testing SeqGeq (pronounced “seek-geek”), desktop software that offers a more user-friendly way for researchers to start asking questions of their data.
“Most biologists were not trained in using software like R,” Hsu said. With SeqGeq, “you are able to play with the data until you can make sense of it and realize how you should move forward.
As the tools become simpler, single-cell research is bound to explode, Mountz said. “This has all kinds of possibilities for cancer, AIDS, Alzheimer’s and a host of other conditions,” he said. “In AIDS, there are CD4 T-cells that are harboring HIV, even after it seems to have been eradicated, and they have the potential to reactivate the infection. How does that work? Everybody has the cells that cause cancer. Why is it that in some people those cells expand and thrive and become pathogenic, whereas in others they are under control? With single-cell sequencing you can study the effects of environment, genes and more on what’s happening inside a person.
“Any disease where there is heterogeneity and a past, present and future state of a cell is a candidate for this approach,” Mountz said. “There’s a whole bunch of low-hanging fruit in every area of science. Today, practically anything you do will be a discovery — it hasn’t been done before at this level. In five years you’ll be repeating what other people have done, but right now we’re at a moment that is wide open.”