Research from UAB published in the New England Journal of Medicine in February 2022 suggests a gene therapy called LentiGlobin could provide a drastic improvement in quality of life for people with sickle cell disease (SCD). Julie Kanter, M.D., director of the UAB Adult Sickle Cell Clinic, says patients treated with this therapy are beginning to show signs of producing stable amounts of normal red blood cells containing hemoglobin.
 Julie Kanter, M.D.
Julie Kanter, M.D.
Sickle cell disease is a group of inherited red blood cell disorders. It is inherited when a child receives two genes—one from each parent—that code for abnormal hemoglobin. Red blood cells contain hemoglobin, a protein that carries oxygen. Healthy red blood cells are round and they move through small blood vessels carrying oxygen to all parts of the body. In a person who has SCD, the hemoglobin is abnormal, which causes the red blood cells to become hard and sticky and look C-shaped, like a sickle farm tool. The sickle cells die early, which causes a constant shortage of red blood cells. Also, when they travel through small blood vessels, they get stuck and clog the blood flow. This can cause pain and other serious complications such as infection, anemia, acute chest syndrome, and stroke.
SCD occurs in about one out of every 365 Black or African American births, according to the Centers for Disease Control and Prevention, and about one in 13 Black or African American babies is born with sickle cell trait.
How It Works
Kanter says there are several types of gene therapy (gene addition/transfer, gene editing, gene correction, and gene silencing), but this particular therapy is categorized as gene addition or transfer.
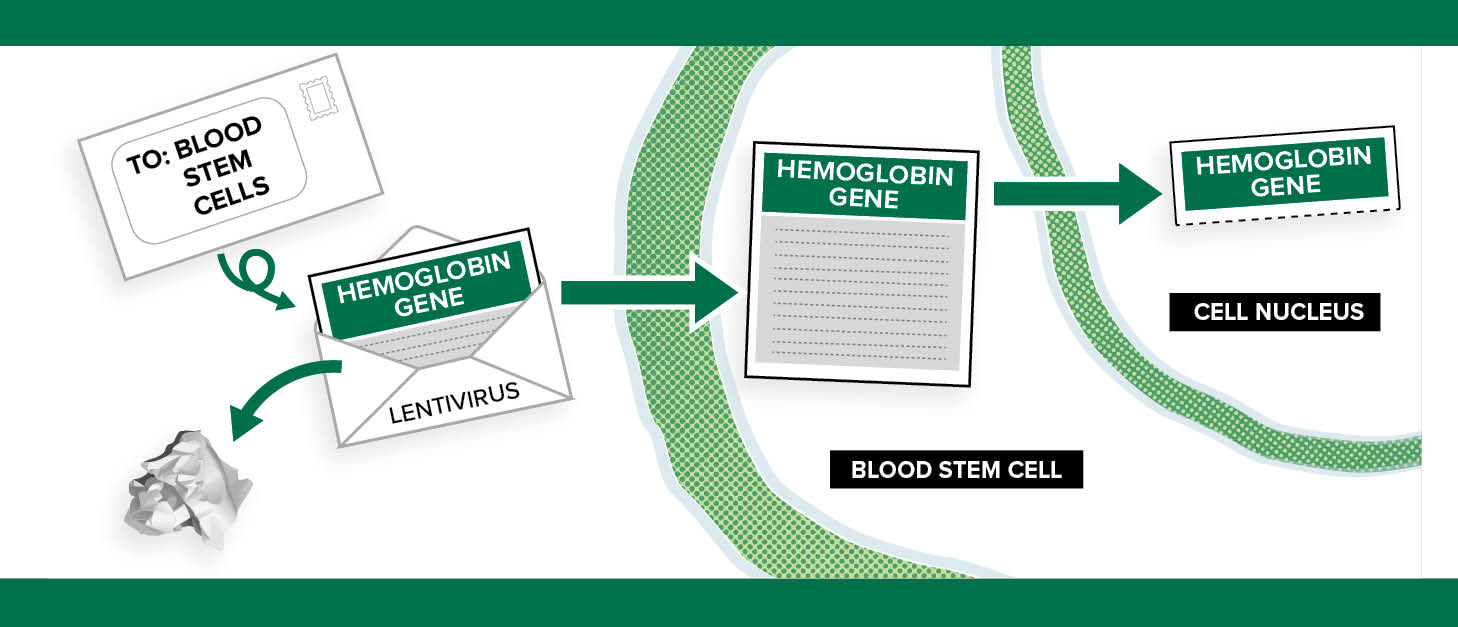 “I like to think of it [the viral vector] as an envelope. We take out the bad part of a virus (the letter) and leave the empty envelope. We put a new gene (the new letter) with the right instructions into the envelope and send it into the stem cells. The viral parts of the letter are removed so patients don’t get the virus itself—they only get the letter coding for the new hemoglobin, called HbAT87Q (a modified type of normal HbA).” —Julie Kanter
“I like to think of it [the viral vector] as an envelope. We take out the bad part of a virus (the letter) and leave the empty envelope. We put a new gene (the new letter) with the right instructions into the envelope and send it into the stem cells. The viral parts of the letter are removed so patients don’t get the virus itself—they only get the letter coding for the new hemoglobin, called HbAT87Q (a modified type of normal HbA).” —Julie Kanter
“In this therapy, we do not change or edit the genes that cause sickle cell disease,” Kanter says. “Instead, we use a viral vector to deliver a new gene that will make a healthy hemoglobin—a beta hemoglobin—into the stem cell. This is like coding new instructions into the cell. The old instructions for the abnormal hemoglobin S (HbS) are still there, but now the cell can make normal HbA (hemoglobin A) and HbS. The vector can deliver more than one copy of the instructions to each cell—usually between one and four copies—so the cell can make more HbA than HbS.”
What Is A Vector?
A vector is part of a virus. Kanter compares vectors to envelopes and letters.
“I like to think of it [the viral vector] as an envelope,” she says. “We take out the bad part of a virus (the letter) and leave the empty envelope. We put a new gene (the new letter) with the right instructions into the envelope and send it into the stem cells. The viral parts of the letter are removed so patients don’t get the virus itself—they only get the letter coding for the new hemoglobin, called HbAT87Q (a modified type of normal HbA).”
What Is T87q?
T87Q is a special type of hemoglobin A that is slightly different from regular hemoglobin A and has two advantages:
-
The intentional change inserted (called T87Q) makes the hemoglobin even less likely to cause sickling when it is near hemoglobin S.
-
The HbAT87Q can also be measured more accurately inside the cell (since it is slightly different from regular HbA), which allows doctors to know how much of the new hemoglobin a patient is making compared to how much they get from a transfusion.
Next Steps
Kanter says that, although this therapy is providing a significant amount of hope, researchers continue to test to make sure the therapy remains safe.
“In an earlier part of this study (now called “group A”) we were not able to get enough of the new gene into each stem cell and we did not have enough total stem cells,” Kanter says. “Not enough envelopes were delivered.”
This caused the stem cells to be extra stressed and the patients to still make red blood cells with HbS and they had symptoms of sickle cell disease. They had only slight improvements in their hemoglobin levels or symptoms (compared to the current group C patients who are doing very well).
“Unfortunately, the stressed-out stem cells may have also housed a bad clone which can cause cancer when isolated in empty bone marrow,” she says. “Two patients in group A developed leukemia because these abnormal cells were too stressed.”
It is important to note that the leukemia was not caused by the viral vector or the new gene (i.e. not from the LentiGlobin) but from the stress of the procedure and the insufficient cell correction.
“We need to see that we have fixed this problem in group C— and that no one else develops leukemia,” Kanter says. “We also need to make sure this procedure both reduces pain/stops all pain crisis and prevents organ damage from sickle cell. This will take time. We will have to watch people for the next two to 15 years and measure their organ function compared to those who did not get this therapy.”
Hope For The Future
Much of Kanter’s career has been dedicated to helping those with SCD. She says a therapy like this is a game changer.
“People with sickle cell disease have endured unnecessary hardship for more than 100 years,” she says. “They have fewer medications and therapies than many other diseases and have received much less attention and funding. We need new and better options for people with sickle cell disease.”
She also says this is just a beginning. “We need to make these treatments available, and we need all people with sickle cell disease to have a sickle cell specialist (doctor) to make that happen. We need the therapy to be affordable so that people everywhere living with this disease have the option for gene therapy. Right now, most people with sickle cell disease live in sub-Saharan Africa and in India. They don’t have even the basic treatments they need like vaccines, penicillin, or hydroxyurea that can make a huge difference in people’s lives with SCD. Eventually we need people in these areas to have equal opportunity to better outcomes.”
 Julie Kanter with a patient at the UAB Adult Sickle Cell Clinic.
Julie Kanter with a patient at the UAB Adult Sickle Cell Clinic.
A Recipe for Success
In August 2022, Kanter was awarded a five-year, $7.7 million grant by the National Institute of Health to help eliminate barriers to care for those with sickle cell disease. She received the grant for her project, “Recruitment and Engagement in Care to Impact Practice Enhancement (RECIPE) for Sickle Cell Disease.” Kanter co-leads the endeavor with several other investigators around the United States. They will use the grant to focus on finding and recruiting people with sickle cell disease (SCD) who are currently unaffiliated from care and understanding their barriers to their care.
Up to 50 percent of affected adults may not see SCD specialists, which limits the delivery of disease-specific screenings and treatment with disease modifying therapies. Issues worsen for individuals living in rural regions or with socioeconomic challenges.
“People not getting the care they need is a major issue for all people with health disparities but especially those with a stigmatizing disease,” Kanter says. “Our goal with this project is to reduce the science-to-practice gap in SCD by identifying individuals who are not receiving guideline-based SCD care.”
Kanter’s project will emphasize the readiness of health systems to serve traditionally underserved populations and sustainability of this work in these areas. The researchers will advance efforts to identify and link unaffiliated patients to SCD specialists by applying implementation science research to adapt existing methods used in human immunodeficiency virus (HIV) care.
“Similar to SCD, individuals with HIV have faced significant healthcare stigma causing reciprocal misgivings about healthcare,” Kanter says. “In this project, we will adapt models for patient identification and engagement in HIV to SCD using a multi-staged, patient-oriented process.”
The Lifespan Comprehensive Sickle Cell Center is part of the Marnix E. Heersink School of Medicine’s Department of Medicine Division of Hematology and Oncology. – Adam Pope