The easy way to carry alignment of multiple, related protein sequencess
Proceed to http://www.expasy.org
In the search box, enter the name of your protein and the species it comes from and press return. Select from the list of records the one that corresponds to your protein (write down the alphanumeric identifying code - you can use that next time you work with the same protein). See an example of this.
At the top right of the list of the selected protein record, press the "Quick BlastP search". This does a BLAST search on the protein database - it's a lot faster than at NCBI.
When the results page for the BLAST search downloads, scroll down to the top of the list of proteins. The first record is the protein you originally selected. The other records are the related proteins to the starting protein in order of their homology scores. Select the ones that are appropriate to compare by checking the box to the left of the record name. The rationale for the selection of these proteins will vary. You may know ahead of time that there are four different isoforms of the protein that you're studying. Or, you may want to compare the same isoform across the species in which it appears.
Once you've made your selections, scroll up to the top of the list of proteins and press the <select> button to the right of the "Send selected sequences to Clustal W" (multiple alignment). This will bring you to the ClustalW XXL page. Your selected sequences should be in the INPUT SEQUENCES box (in FASTA format). Enter your e-mail address and then press the "Run ClustalW" button.
This will very rapidly return the ClustalW-XXL query receipt. There are two outputs - Multiple alignments and Dendrograms. Select ClustalW (aln) to download the aligned sequences. They are in order of how you selected them.
Underneath the line of stack sequences, there is an indication of the strength of the homology - perfectly homologous residues are designed with an asterisk*. Closely related residues such as L/I/V are noted by a colon ":", whereas less homologous residues such as A/G, A/V, P/S, N/S are noted by a period "."
Copy the entire page and put it into Microsoft Word. Select all the transferred text (control-A) and convert it the Courier New font. This font uses equal spacing of the letters and that will ensure that the alignment is properly sustained. You may need to change the font size and/or the page width so that sequences are not broken up.
A useful feature of Word is that by pressing the Option key, you can select/highlight (vertically) the corresponding amino acid in each record. Once selected, you can go to the highlighting toolbar and color code that vertical column.
Example of multiply aligned sequences.
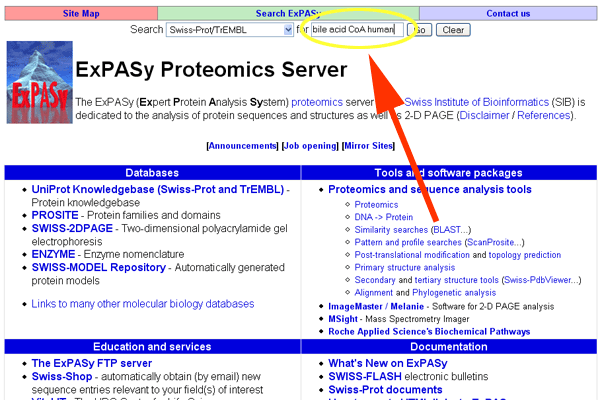
- Go to http://www.expasy.org. Enter "bile acid CoA human".
See larger image.
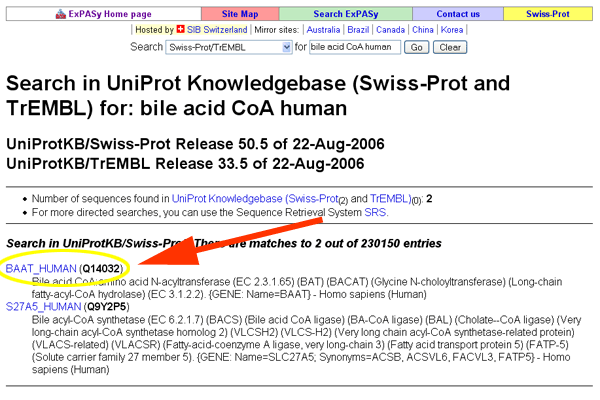
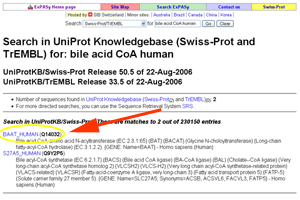
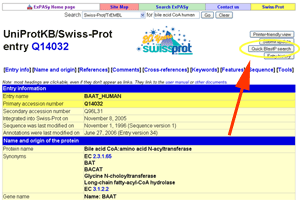
- This will bring up a record for BAAT-HUMAN (Q14032).
See larger image.
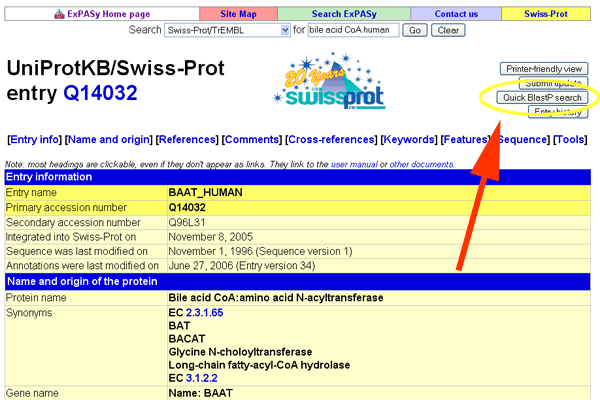
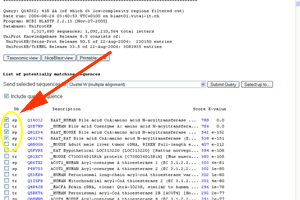
- Do a BLASTP search.
See larger image.
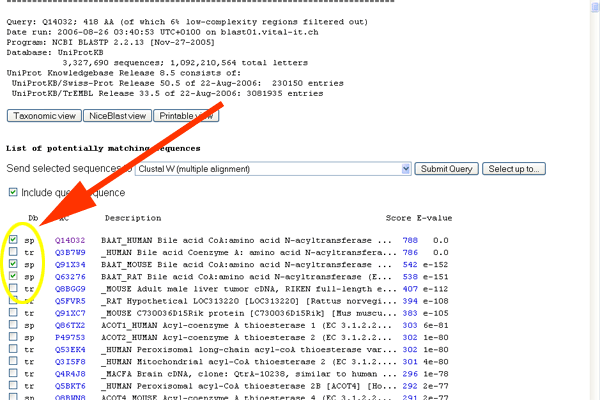
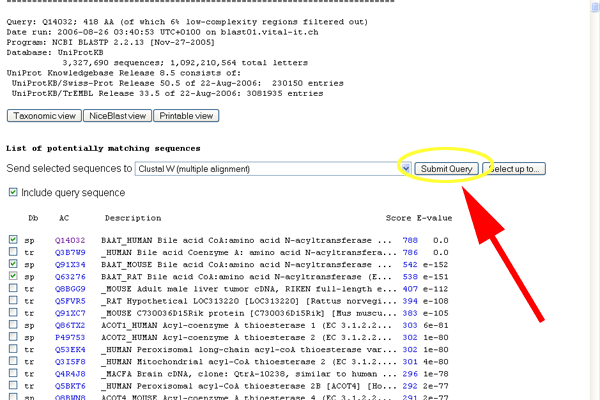
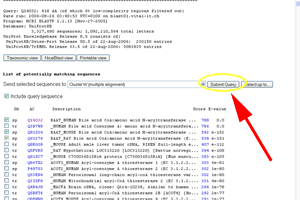
- Check the boxes for Q14032, Q91X34 and Q63276 - these are the sequences for the human, mouse and rat version of BAAT.
See larger image.
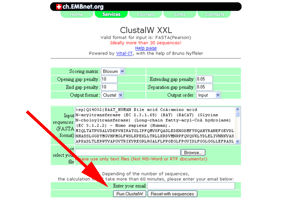
- Press the submit button for the ClustalW search.
See larger image.
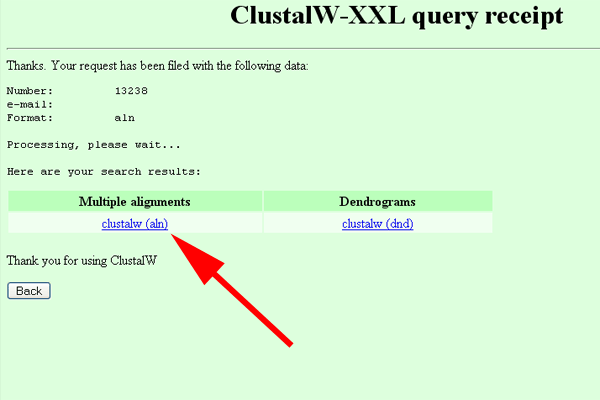
- At the ClustalW-XXL page, press the "Run ClustalW" button.
See larger image.
- On the ClustalW-XXL query receipt page, click on "clustalw (aln)".
See larger image.
- The results can be transferred to Word files, either with no color coding or with homologies enhanced by color highlighting.