 Steven Pittler, Ph.D.Retinal degeneration in the disease retinitis pigmentosa is caused by a family of hereditary mutations that slowly lead to blindness over years or decades. A mouse model of one of these forms of retinitis pigmentosa, RP59, exhibits key biochemical and diagnostic features of human RP59, University of Alabama at Birmingham researchers report in the journal Cell Death and Disease.
Steven Pittler, Ph.D.Retinal degeneration in the disease retinitis pigmentosa is caused by a family of hereditary mutations that slowly lead to blindness over years or decades. A mouse model of one of these forms of retinitis pigmentosa, RP59, exhibits key biochemical and diagnostic features of human RP59, University of Alabama at Birmingham researchers report in the journal Cell Death and Disease.
“Further, we show structural and inner retina functional deficits in the K42E mouse model that are consistent with altered synaptic transmission between retinal photoreceptors and bipolar cells,” said Steven Pittler, Ph.D., UAB leader of the research. “This investigation may provide novel insights into RP59 disease mechanism that will guide future testing of therapeutic interventions to slow or halt the progression of this blinding disorder.”
RP59 is caused by the mutation of one amino acid in an enzyme called DHDDS that is part of an enzyme complex involved in the glycosylation of proteins. The K42E mouse model replicates this mutation by changing the same amino acid in the mouse enzyme, from lysine to glutamic acid. Pittler and colleagues in the UAB Department of Optometry and Vision Science and other institutions first reported the mouse model in 2020, and preliminary study showed lack of profound retinal degeneration and protein N-glycosylation defects.
Their current study now presents a detailed report of the inner retina pathology and defective synaptic transmission caused by this recessive homozygous mutation.
The enzyme complex that contains DHDDS catalyzes the stepwise addition of five-carbon isoprene units to elongate a chain of polyprenyl diphosphate that is later transformed to dolichol, the long-chain organic compound that is required for protein N-glycosylation. In normal human bodily tissues and fluids, the predominant strain of dolichol contains 19 isoprene units. In human RP59 patients, dolichol length shifts to 18 or 17 isoprene units, which is diagnostic for RP59.
UAB researchers found that, in the K42E mouse model, dolichol species in the retina, liver and brain were also shortened, similar to RP59.
Transcriptomic analysis of neurons in the retina revealed that 68 genes were differentially expressed in the K42E mice compared to wild-type controls — 54 up-regulated and 14 down-regulated. Because many of these differentially regulated genes are involved in generation of synapses, which are the sites of electric nerve impulses between two nerve cells, and in function of the synapses, the researchers took a closer look at the structure and function of the inner and outer retina in the K42E mice.
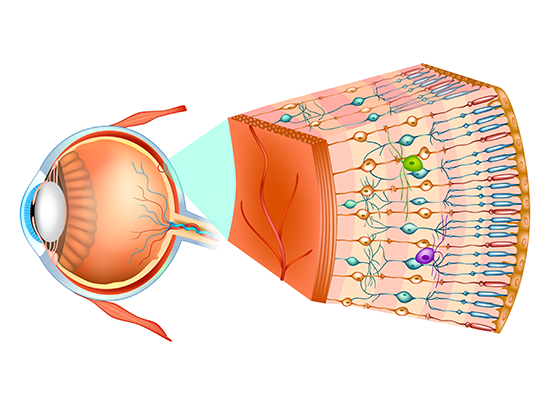 The model may provide novel insights into RP59 disease mechanism that will guide future testing of therapeutic interventions.The retina has 10 distinct layers of neurons that are interconnected by synapses. Nerve signals are generated in the cells that detect light and then pass through successive layers of neurons that process the signals and then send that information through the optic nerve to the visual cortex of the brain. Detailed measurements of retinal cell layer thickness in K42E mice revealed a significant reduction of one of the neural layers, the inner nuclear layer, as well as a small reduction in total retinal thickness. This thinning begins two months after birth, and it progressively increases through 18 months after birth.
The model may provide novel insights into RP59 disease mechanism that will guide future testing of therapeutic interventions.The retina has 10 distinct layers of neurons that are interconnected by synapses. Nerve signals are generated in the cells that detect light and then pass through successive layers of neurons that process the signals and then send that information through the optic nerve to the visual cortex of the brain. Detailed measurements of retinal cell layer thickness in K42E mice revealed a significant reduction of one of the neural layers, the inner nuclear layer, as well as a small reduction in total retinal thickness. This thinning begins two months after birth, and it progressively increases through 18 months after birth.
Microscopic study revealed cell loss in the inner nuclear layer, disruption in the outer plexiform layer, infiltration of photoreceptor nuclei into the outer plexiform layer and pronounced retraction of photoreceptor terminals into the outer nuclear layer. There was also evidence of slow cell death of retinal neurons, beginning one month after birth.
The UAB researchers used electroretinography — where an electrode on the surface of the eye measures the electrical responses of retinal neurons to a flash of light — to determine which layers of retinal neurons were defective. The first electrical response, called the a-wave, reflects the health of the photoreceptor cells in the outer retina that detect photons. The second response, the b-wave, reflects the health of the inner layers of the retina, which lie downstream from the photoreceptor cells.
Researchers found that the K42E mice had reduced b-wave amplitudes that began one month after birth and progressively declined through 18 months after birth, without appreciable a-wave attenuation.
“Our results suggest that the underlying cause of DHDDS K42E variant-driven RP59 retinal pathology is defective synaptic transmission from outer to inner retina,” Pittler said.
Co-first authors of the study, “A Dhdds K42E knock-in RP59 mouse model shows inner retina pathology and defective synaptic transmission,” are Mai N. Nguyen and Dibyendu Chakraborty, UAB Department of Optometry and Vision Science.
Other co-authors along with Pittler, Nguyen and Chakraborty are Sriganesh Ramachandra Rao and Steven J. Fliesler, VA Western New York Healthcare System and the University at Buffalo, Buffalo, New York; Agnieszka Onysk, Mariusz Radkiewicz, Liliana Surmacz and Ewa Swiezewska, Polish Academy of Sciences, Warsaw, Poland; Eric Soubeyrand and Tariq A. Akhtar, University of Guelph, Guelph, Ontario, Canada; Timothy W. Kraft, UAB Department of Optometry and Vision Science; and David M. Sherry, University of Oklahoma Health Sciences Center, Oklahoma City, Oklahoma.
Support came from National Institutes of Health grants EY029341, EY003039 and EY032764; a Fight for Sight Summer Student Fellowship; a Career-Starter Research Grant from the Knights Templar Eye Foundation; and National Science Centre of Poland grant UMO-2019/35/B/NZ1/03794.
Optometry and Vision Science is the department in the UAB School of Optometry, and Pittler is director of the Vision Science Research Center, also in the School of Optometry.