Media contact: Bob Shepard
 Steven RoweLong before researchers found the cause of cystic fibrosis — a single gene on the long arm of chromosome 7 — this inherited disease was often diagnosed with a kiss. The taste of super-salty sweat on their babies’ skin alarmed parents and sent them to their doctors. Doctors, by the early 1950s, realized that abnormal sweat was a hallmark of a disease they soon defined as cystic fibrosis, a disease so severe that most patients would die in early childhood.
Steven RoweLong before researchers found the cause of cystic fibrosis — a single gene on the long arm of chromosome 7 — this inherited disease was often diagnosed with a kiss. The taste of super-salty sweat on their babies’ skin alarmed parents and sent them to their doctors. Doctors, by the early 1950s, realized that abnormal sweat was a hallmark of a disease they soon defined as cystic fibrosis, a disease so severe that most patients would die in early childhood.
The trouble with mucus
Unless you have a cold, you probably don’t give mucus a second thought. But people with cystic fibrosis can often think of little else. Thick, sticky mucus coats their airways and clogs their lungs, making breathing a chore and infections a constant threat. Salty sweat is another symptom of the same root problem: one of the body’s key hydration regulators is broken. At the dawn of the 1960s, airway clearance exercises and aggressive and frequent therapy with powerful antibiotics began to push the average lifespan higher. But children with CF like Gregory Fleming James, second son of eventual Alabama governor Fob James, still didn’t generally live past 10. Gregory didn’t even make that milestone; he died in 1967, aged 8.
This animation illustrates what happens when CFTR protein channels malfunction in cystic fibrosis. Video courtesy Cystic Fibrosis Foundation.
'The clock was always ticking'
By the early 1990s, despite new, more powerful antibiotics and the use of transplants to replace failing lungs, the average lifespan for a young adult still hovered around age 20. In those years, Steven Rowe, M.D., was a teenage counselor at a camp for children with special medical needs. He got to know “the trials and lack of effective treatments” for people with cystic fibrosis first hand. “They had to have hours of physiotherapy every day, and even if they did everything right, the disease would continue its relentless progress,” Rowe recalls. In 1989, researchers had traced the cause of cystic fibrosis to mutations in the CFTR gene, and they thought a gene therapy-based cure might be just around the corner. But the day-to-day lives of patients hadn’t much changed, says Rowe. “The clock was always ticking.”
A new kind of research center
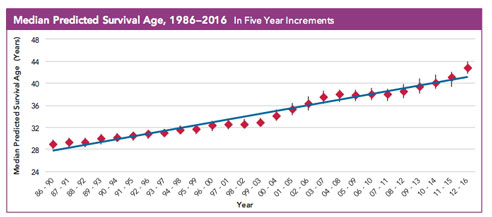 Survival rates have been climbing steadily in cystic fibrosis for decades. Image courtesy Cystic Fibrosis Foundation.When he came to UAB for his medical residency in 1998, Rowe was determined to find treatments that could put time back on that clock. He joined internationally recognized scientists such as Eric Sorscher, Ph.D., and J.P. Clancy, M.D., at UAB’s Gregory Fleming James Cystic Fibrosis Research Center, which had been established in 1981 during Fob James’ third year as governor. The UAB CF Center, which Rowe now directs, was one of the first such centers in the country, and it has been continually funded by the NIH and the Cystic Fibrosis Foundation ever since. The center’s basic and translational research discoveries have helped transform the disease. Today, the average lifespan for a patient with cystic fibrosis is 47 and climbing fast. Rowe now can offer a growing number of his patients highly effective drug therapies that can clear their mucus blockages, improve breathing and dramatically improve their quality of life, almost overnight.
Survival rates have been climbing steadily in cystic fibrosis for decades. Image courtesy Cystic Fibrosis Foundation.When he came to UAB for his medical residency in 1998, Rowe was determined to find treatments that could put time back on that clock. He joined internationally recognized scientists such as Eric Sorscher, Ph.D., and J.P. Clancy, M.D., at UAB’s Gregory Fleming James Cystic Fibrosis Research Center, which had been established in 1981 during Fob James’ third year as governor. The UAB CF Center, which Rowe now directs, was one of the first such centers in the country, and it has been continually funded by the NIH and the Cystic Fibrosis Foundation ever since. The center’s basic and translational research discoveries have helped transform the disease. Today, the average lifespan for a patient with cystic fibrosis is 47 and climbing fast. Rowe now can offer a growing number of his patients highly effective drug therapies that can clear their mucus blockages, improve breathing and dramatically improve their quality of life, almost overnight.
'One of the most remarkable stories in modern medicine'
“This is one of the most remarkable stories in modern medicine,” says Rowe, professor in the Division of Pulmonary, Allergy and Critical Medicine in the UAB School of Medicine and Nancy R. and Eugene C. Gwaltney Family Endowed Chair in Medical Research at UAB. In CF, genetic testing is now almost universal and mutation-based drug prescribing is standard practice. That offers a template for other diseases looking to translate the promise of precision medicine into the daily lives of patients, Rowe notes. (Matt Might, Ph.D., director of UAB’s Hugh Kaul Precision Medicine Institute, says Rowe and the CF Center were a major factor that attracted him to Birmingham.) More than once, UAB researchers pursued high-risk strategies that many in the field thought were bound to fail. Those efforts have already reaped major dividends, Rowe says.
Highly effective therapies that target the basic defect should provide therapeutic benefits for 90 percent of patients with cystic fibrosis, according to a study published by Rowe and colleagues in the New England Journal of Medicine in October 2018. And UAB researchers are exploring promising new ways to reach even those with the rarest of CF mutations. Meanwhile, they’re applying CF breakthroughs to other lung diseases, from COPD to asthma and beyond. Advances that were initially designed for only a few thousand people with cystic fibrosis could eventually reach tens of millions more.
Here's how:
How did we get here? Cystic fibrosis drugs go from 0-90 percent effective in a few short years
Despite early hopes for gene therapy cures for cystic fibrosis, “the field underestimated how difficult it would be to make them work,” explains Steven Rowe, M.D., director of the UAB Gregory Fleming James Cystic Fibrosis Research Center. (Gene therapy approaches are still being studied and have potential, he notes.) “It was only when we said, ‘Let’s try to fix the protein rather than the gene,’ that we had this big uplift in therapeutic success.”
Toward protein-based treatment
Fixing a protein, unlike fixing a gene, can mean correcting a whole series of errors. The CFTR protein is a gatekeeper. Its station is in the cell membrane, where it allows a variety of substances, including chloride, bicarbonate and ultimately water, to pass in and out of the cell, and acts as a “master regulator of hydration status,” says George Solomon, M.D., a researcher in the UAB CF Center and assistant professor in the Division of Pulmonary, Allergy and Critical Medicine in the UAB School of Medicine. There are plenty of ways it can go wrong. Just short of 2,000 mutations have been documented in the CFTR gene, which is the template for making the CFTR protein; hundreds of those mutations are known to lead to CF symptoms.
'Very high-risk'
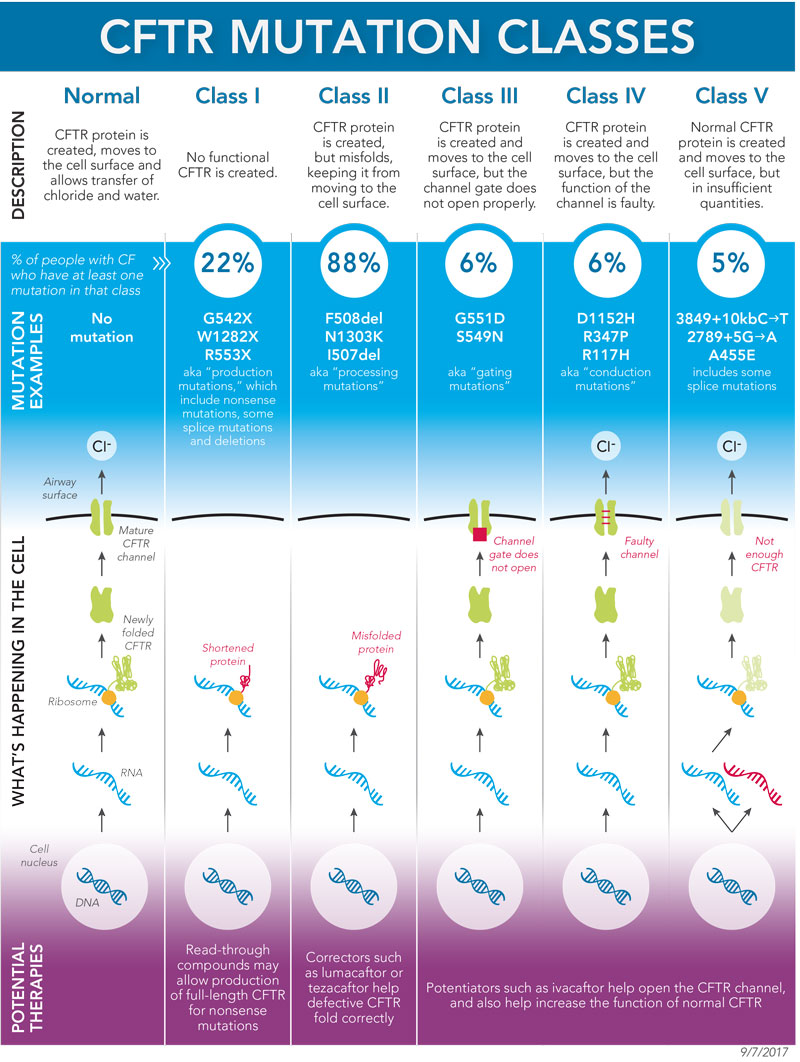 This graphic from the Cystic Fibrosis Foundation illustrates normal CFTR functioning (far left) and the different types of mutation classes in cystic fibrosis. Image courtesy Cystic Fibrosis Foundation.The end result is generally the same: dehydrated airways and deranged mucus. But the specific cause in any individual case goes back to the mutation. Some patients have mutations that affect the CFTR channel itself, preventing it from opening properly. Other mutations cause the CFTR proteins to become misfolded, so that they never even reach the cell membrane. With all this molecular chaos, could a protein-based treatment ever work? Many in the field were doubtful. The focus on protein repair “was considered very high-risk at the time,” Rowe says. “But it has paid dividends.”
This graphic from the Cystic Fibrosis Foundation illustrates normal CFTR functioning (far left) and the different types of mutation classes in cystic fibrosis. Image courtesy Cystic Fibrosis Foundation.The end result is generally the same: dehydrated airways and deranged mucus. But the specific cause in any individual case goes back to the mutation. Some patients have mutations that affect the CFTR channel itself, preventing it from opening properly. Other mutations cause the CFTR proteins to become misfolded, so that they never even reach the cell membrane. With all this molecular chaos, could a protein-based treatment ever work? Many in the field were doubtful. The focus on protein repair “was considered very high-risk at the time,” Rowe says. “But it has paid dividends.”
Groundbreaking trials
And how. When pharmaceutical company Vertex found promising drug candidates, tests developed in Rowe’s lab helped home in on the compounds most likely to improve CFTR protein function. UAB patients were some of the earliest enrollees in the groundbreaking trials of Vertex’s drug ivacaftor, the first protein-focused treatment for cystic fibrosis, which received FDA approval in 2012. “The first time we treated patients with ivacaftor,” in a clinical trial a few years earlier, “We got a call from one patient within a couple of days,” Rowe says. “She said, ‘I’m finally coughing up all this mucus.’ We knew something good might be happening.”
After a lifetime of needing family members to pound out their recalcitrant mucus, patients were getting rid of it on their own. They could breathe more easily. Follow-up studies found that patients could fight off seemingly fatal infections when they were on ivacaftor. Children were growing bigger and stronger. “Ivacaftor is like a modern-day miracle drug for some patients,” says Jennifer Guimbellot, M.D., Ph.D., a pediatric pulmonologist, assistant professor in the Department of Pediatrics and researcher in the UAB CF Center. “I’ve seen remarkable improvements in my patients.”
UAB researcher George Solomon explains advances in CF therapies in this video from the Cystic Fibrosis Foundation. Video courtesy Cystic Fibrosis Foundation.
Beyond gating mutations
But ivacaftor was always only the tip of the iceberg. A mere 4-6 percent of CF patients have the “gating” mutations that ivacaftor targets, in which CFTR channels cannot open. For the vast majority of patients, the problem is misfolded proteins. Still, the success of ivacaftor “brought a lot of others into the field,” Rowe says.
Correctors, a new class of drugs that help proteins fold properly, arrived a few years ago. They were only partially effective on their own, says Rowe. But in major multi-center trials that he co-led, combinations of correctors and ivacaftor have brought significant improvement. Currently, some 15 percent of patients have highly effective CFTR-directed therapies available, and another 50 percent also benefit from these potentiator-corrector combinations.
The triple combo
The next step is the triple combination. Rowe is now co-leading large studies of drugs that combine two correctors plus ivacaftor, to bring even more channels to the membrane. Results from phase 2 trials, reported in October in the New England Journal of Medicine, have been just as impressive as the original ivacaftor studies, he says, including “major improvements in lung function and quality of life.” Even the sweat test, the harbinger of bad things known to the taste buds of mothers and infants, began to normalize. If these responses continue in phase 3 trials, up to 90 percent of CF patients will have highly effective CFTR therapy.
Even seemingly minor improvements can make a major difference, Rowe notes. In a recent commentary in Lancet Respiratory Medicine, he explains that reducing the average rate of decline in a 20-year-old patient by a single percentage point, from 4 percent to 3 percent per year, would add an extra 6-8 years of life. “We’re on the brink of seeing this through from vision to execution and availability,” he says.
Attacking nonsense mutations in cystic fibrosis and a host of other diseases
 David BedwellFor the 5-10 percent of cystic fibrosis patients with “nonsense” mutations, the trouble isn’t defective or misfolded proteins — it’s no protein at all. Somewhere in their CFTR genes, a three-letter code that should tell the cell’s protein-building ribosomes to insert an amino acid has been accidentally changed into a premature “stop” sign. A one-letter switch can turn, for example, UAC (code for tyrosine) into UAA (which tells the ribosome, “stop, you’ve finished making this protein”). Because the protein is foreshortened and clearly abnormal, it is quickly destroyed by the cell’s quality-control processes.
David BedwellFor the 5-10 percent of cystic fibrosis patients with “nonsense” mutations, the trouble isn’t defective or misfolded proteins — it’s no protein at all. Somewhere in their CFTR genes, a three-letter code that should tell the cell’s protein-building ribosomes to insert an amino acid has been accidentally changed into a premature “stop” sign. A one-letter switch can turn, for example, UAC (code for tyrosine) into UAA (which tells the ribosome, “stop, you’ve finished making this protein”). Because the protein is foreshortened and clearly abnormal, it is quickly destroyed by the cell’s quality-control processes.
The readthrough breakthrough
That’s why patients with nonsense mutations usually have a severe form of the disease, explains David Bedwell, Ph.D., associate director of the UAB Gregory Fleming James Cystic Fibrosis Research Center and chair of the Department of Biochemistry and Molecular Genetics in the UAB School of Medicine. In 1996, Bedwell and colleagues Marybeth Howard, Ph.D., and former CF Center director Raymond Frizzell, Ph.D., published a paper in Nature Medicine that proposed a way around the problem. Some antibiotic drugs, they showed, could encourage ribosomes to “read through” the premature stop signs and go on to make functional protein. Readthrough drugs, they proposed, could be a new treatment option for CF.
“Right after we published that, someone wrote a letter to the editor saying it would never work,” says Bedwell. It sounds like a “crazy idea,” he admits, but it is beginning to prove itself in patients. In Europe, a drug Bedwell has studied extensively, ataluren, is now used to treat Duchenne’s muscular dystrophy, and Bedwell is exploring promising results for ataluren in several other diseases, including Hurler syndrome, also known as mucopolysaccharidosis type 1. (Matt Might, Ph.D., through the UAB Precision Medicine Institute, is now attracting patients with many other genetic diseases caused by nonsense mutations; these diseases may also be future targets to readthrough therapies.)
This video from the Cystic Fibrosis Foundation explains what goes wrong in nonsense mutations and gives an overview of research efforts to correct them. Video courtesy Cystic Fibrosis Foundation.
Overriding the proofing function
The trick, Bedwell says, is to make the cell’s proofreading mechanisms a little more lenient. Occasionally, the protein-building ribosomes accidentally grab the wrong base as they work. Confronted with UAA (stop!), the ribosome might latch on to cytosine (C) instead of uracil (U), ending up with CAA (glutamine) instead. Now there’s no stop sign, and the ribosome keeps on building CFTR. (Even though CAA is the wrong amino acid, such a small variation doesn’t usually affect the protein’s function.) The ribosome’s proofing function normally catches and fixes these deviations from the written code, meaning that patients don’t get to benefit from the ribosome’s error. But the presence of a drug like ataluren seems to encourage the ribosome to ignore the mistake and go on making the protein.
Unfortunately, the process still relies on chance, and current readthrough drugs can’t boost protein production to the 30-35 percent levels needed to make a major difference in CF, Bedwell explains. So he and partners at UAB-affiliate Southern Research and the Cystic Fibrosis Foundation are seeking a better compound. The team has sifted through 750,000 options and is homing in on the 150-or-so most promising hits.
International effort
Because nonsense mutations are highly varied, it is important to have a range of samples to test potential new drugs. These will be available at UAB thanks to a multinational study led by George Solomon, M.D., a researcher in the CF Center and assistant professor in the Division of Pulmonary, Allergy and Critical Medicine. The RARE study is collecting a selection of tissue samples from patients with nonsense mutations in several countries to learn more about the biology of these mutations and to allow for next-generation cell-based tests, Solomon says.
A new drug could have uses far beyond CF, Bedwell points out. Up to 11 percent of all genetic diseases are thought to be caused by nonsense mutations, he says. “These drugs could help millions worldwide.”
Clinical trial for one: the promise of patient-derived assays
 Jennifer GuimbellotEven as new drug combinations bring effective therapy to more patients with cystic fibrosis, the sheer volume of CF mutations means many patients have little hope of seeing a drug company focus on their particular genetic problem.
Jennifer GuimbellotEven as new drug combinations bring effective therapy to more patients with cystic fibrosis, the sheer volume of CF mutations means many patients have little hope of seeing a drug company focus on their particular genetic problem.
Ivacaftor brought remarkable improvements to many of her patients, says Jennifer Guimbellot, M.D., Ph.D., a pediatric pulmonologist, assistant professor in the Department of Pediatrics in the UAB School of Medicine and a researcher in the UAB Gregory Fleming James Cystic Fibrosis Research Center. But children with rare mutations still have few good options when they fall ill.
“It’s tough to watch,” Guimbellot says, recalling two patients who barely survived harrowing exacerbations. “The parents were accepting, saying, ‘These drugs are not for my kid, and maybe they never will be.’ But I thought it was really unfair.”
Helping the 'lost patients'
“Those are the ‘lost patients’ in CF,” says George Solomon, M.D., a researcher in the CF Center and assistant professor in the Division of Pulmonary, Allergy and Critical Medicine. “There may never be a study that targets their mutation specifically.” The mutations in CF are so varied, the mutations so rare, and the drugs so new, that it’s often impossible to judge whether a drug will be effective in a patient with a rare mutation. And with the cost of drugs such as ivacaftor surpassing $350,000 per year, insurance companies want to see evidence to suppose a drug may work in a particular patient before they approve reimbursement.
 George SolomonThe answer, Guimbellot and Solomon both say, is to develop assays from a patient’s cells that would let physicians predict that patient’s response in the lab — a personal clinical trial that can be run any time it’s needed. Solomon’s approach, using air-liquid interface cultures, has already demonstrated the viability of the combination drug Orkambi for one young patient with a rare mutation, who was referred to UAB from a hospital in New England. “They knew we were working on this, so they sent him here,” Solomon says. His tests showed Orkambi could help, and the patient’s insurance company agreed to pay for treatment. “Two years later, he’s still on the drug and doing well.”
George SolomonThe answer, Guimbellot and Solomon both say, is to develop assays from a patient’s cells that would let physicians predict that patient’s response in the lab — a personal clinical trial that can be run any time it’s needed. Solomon’s approach, using air-liquid interface cultures, has already demonstrated the viability of the combination drug Orkambi for one young patient with a rare mutation, who was referred to UAB from a hospital in New England. “They knew we were working on this, so they sent him here,” Solomon says. His tests showed Orkambi could help, and the patient’s insurance company agreed to pay for treatment. “Two years later, he’s still on the drug and doing well.”
The smart tubes
Guimbellot’s approach, which she calls nasospheroids, involves epithelial cells harvested from a patient’s nose. The cells naturally want to form a tube — “I tell patients, ‘they like to have friends,’” she says — and in a fluid-filled lab dish they spontaneously link up in a ring shape, about the diameter of a human hair, with liquid in the center. She first encountered this phenomenon in a lab at the University of North Carolina – Chapel Hill. “As soon as I saw that, I said, ‘This is something I can use,’” Guimbellot recalls.
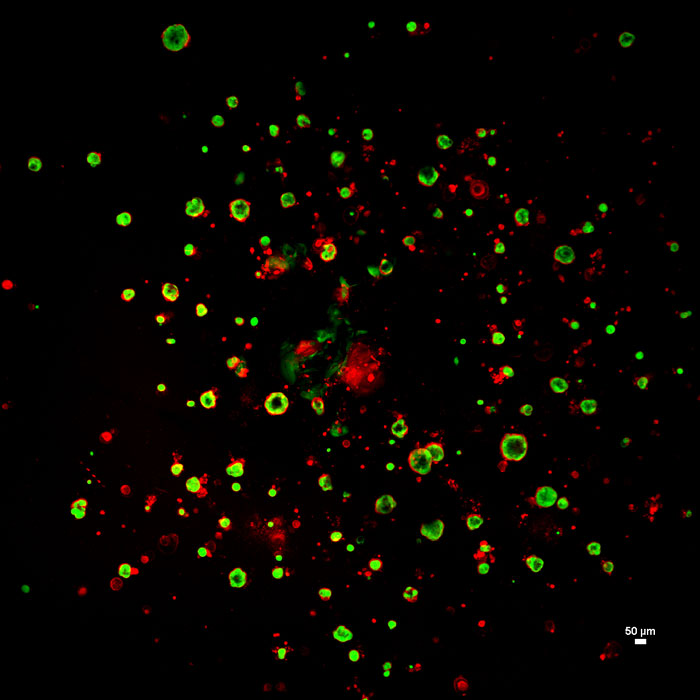 Living nasospheroids derived from nasal cells from a patient with cystic fibrosis. The cells are stained with the fluorescent dyes calcein green and plasma membrane orange for imaging at 4x magnification. Guimbellot calls these nasospheroids AMIs, for "apical membrane in" — she also makes AMOs, or "apical membrane out" — referring to the side of the nasospheroid facing what would be the airway in the patient's body. Image courtesy Jennifer Guimbellot.
Living nasospheroids derived from nasal cells from a patient with cystic fibrosis. The cells are stained with the fluorescent dyes calcein green and plasma membrane orange for imaging at 4x magnification. Guimbellot calls these nasospheroids AMIs, for "apical membrane in" — she also makes AMOs, or "apical membrane out" — referring to the side of the nasospheroid facing what would be the airway in the patient's body. Image courtesy Jennifer Guimbellot.
The video below shows swirling mucus and fluid inside a nasospheroid derived from nasal cells from a non-CF patient, imaged at 40x magnification. Nasospheroids derived from CF patients show little mucus and fluid movement due to dysfunctional CFTR. Video courtesy Jennifer Guimbellot.
Cells taken from healthy volunteers, with functioning CFTR channels, will move chloride in or out of the ring, and water naturally follows. In healthy people, the ring changes size based on the activity of CFTR. Cells from CF patients, lacking functional CFTR channels, don’t change size — until Guimbellot adds ivacaftor. The amount of change may be a sensitive guide to how well a patient will respond to the drug, Guimbellot has found. Especially as more new CF drug combinations reach the clinic, “I want to know if we can use these cell assays will tell me whether a patient will respond or not,” Guimbellot says.
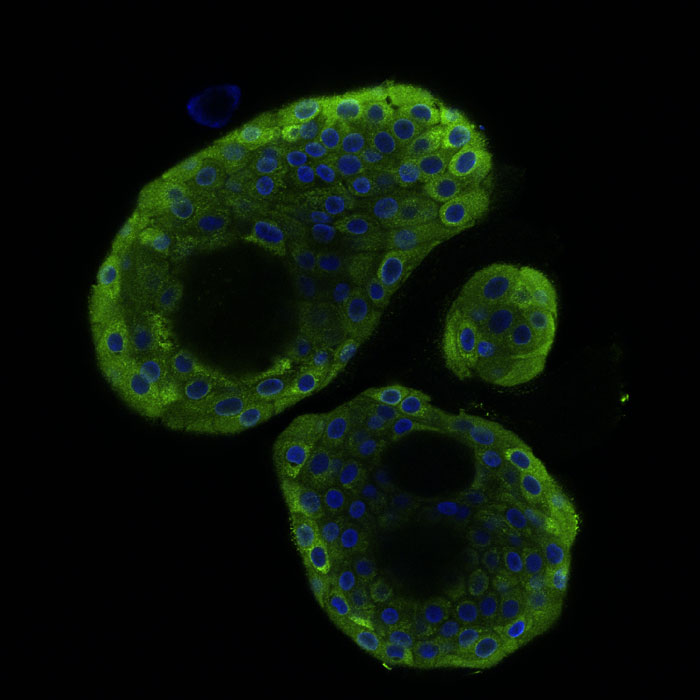 Nasospheroids derived from nasal cells from a patient with cystic fibrosis. They are fixed (non-living), and stained with antibodies to CFTR (green areas in the image), the protein that is dysfunctional in CF patients, and Nucblue, a dye for the nucleus, at 40x magnification. CFTR is normally localized on the inner membrane, but in this CF patient the CFTR is distributed throughout the cells due to the dysfunctional localization of CFTR. Image courtesy Jennifer Guimbellot.
Nasospheroids derived from nasal cells from a patient with cystic fibrosis. They are fixed (non-living), and stained with antibodies to CFTR (green areas in the image), the protein that is dysfunctional in CF patients, and Nucblue, a dye for the nucleus, at 40x magnification. CFTR is normally localized on the inner membrane, but in this CF patient the CFTR is distributed throughout the cells due to the dysfunctional localization of CFTR. Image courtesy Jennifer Guimbellot.
Nasospheroids could also help in patients with well-known mutations, Guimbellot adds. As future generations of drugs reach the clinic, these little spheroids, or their successors, could quickly tell her if any of her patients could benefit by switching to the new treatments. And she isn’t focused solely on CFTR. That’s because mutations in other genes, ones involved in drug metabolism, have an effect on how well treatments affect individual patients. The cytochrome P450 enzyme family, for instance, breaks down many types of drugs. And that means patients with active P450 enzymes may need higher doses to achieve the same effect, she says.
“You can see 20 patients with the exact same [CFTR] mutation, and they have different responses to the same drug,” Guimbellot says. “I envision a time when everyone will start on the right drug at the right dose, instead of relying on trial and error.”
From cystic fibrosis to COPD: potentiators and chronic bronchitis
 Mark DransfieldCystic fibrosis and chronic bronchitis share many similarities. Both are marked by mucus obstruction of the small airways and daily production of sputum. What differs is how many people they affect: while 30,000 Americans have CF, millions have chronic bronchitis, which is caused largely by smoking and is a major cause of chronic obstructive pulmonary disease (COPD). COPD is one of the world’s leading causes of death, “and it’s the only one in the top 10 that’s growing in the United States,” notes Steven Rowe, M.D., director of the UAB Gregory Fleming James Cystic Fibrosis Research Center.
Mark DransfieldCystic fibrosis and chronic bronchitis share many similarities. Both are marked by mucus obstruction of the small airways and daily production of sputum. What differs is how many people they affect: while 30,000 Americans have CF, millions have chronic bronchitis, which is caused largely by smoking and is a major cause of chronic obstructive pulmonary disease (COPD). COPD is one of the world’s leading causes of death, “and it’s the only one in the top 10 that’s growing in the United States,” notes Steven Rowe, M.D., director of the UAB Gregory Fleming James Cystic Fibrosis Research Center.
What other patients could benefit?
Once researchers saw how well ivacaftor cleared mucus in CF patients, “we started to think, What other patients could benefit?” Rowe says. Chronic bronchitis was at the top of the list. In his lab, Rowe found that exposing healthy airway cells to cigarette smoke reduced CFTR activity, and that CFTR proteins were present at half the normal level in cells taken from smokers.
A small trial in UAB’s Lung Health Center confirmed those findings, and established a partnership between Rowe’s team and Mark Dransfield, M.D., medical director of the Lung Health Center. CFTR activity was lower than normal in the noses and lungs of patients with chronic bronchitis, Dransfield says. “It seems to be an intermediate phenotype — not as dysfunctional as in CF, but definitely abnormal.” That led to a pilot study in which patients with chronic bronchitis received ivacaftor. Results were promising, and a larger trial is now underway. “In our anecdotal experience, we’ve had some pretty dramatic responses,” Dransfield says. “Some people are saying they have no sputum production, that they feel great.” In a separate larger study reported this year by Rowe with a different compound that potentiates CFTR like ivacaftor, the investigational medicine improved lung function and markers of inflammation, and will be brought forward in much larger studies.
No good therapies
Chronic bronchitis, like CF a decade ago, has no therapies that get to the root of the problem, Dransfield notes. Alabama is particularly hard-hit, ranking second in the nation in chronic bronchitis prevalence. “The hope is this would be the first efficacious treatment” for mucus-clogged patients, Dransfield says. “We’re hoping to apply a drug developed for a few thousand people with cystic fibrosis to up to 10 million people with chronic bronchitis.”
Muco-vision: a new imaging approach for cystic fibrosis, asthma and beyond
Several imaging devices let researchers focus on mucus in airways, the epithelial cells that line those airways, or the tiny, hair-like cilia swaying on the outside of the epithelial cells. Only one technology — a joint invention of UAB researchers and investigators at Harvard Medical School — lets them observe all three at once. Known as micro-optical coherence tomography, or microOCT, it is “basically like ultrasound with a laser,” says Steven Rowe, M.D., director of the UAB Gregory Fleming James Cystic Fibrosis Research Center.
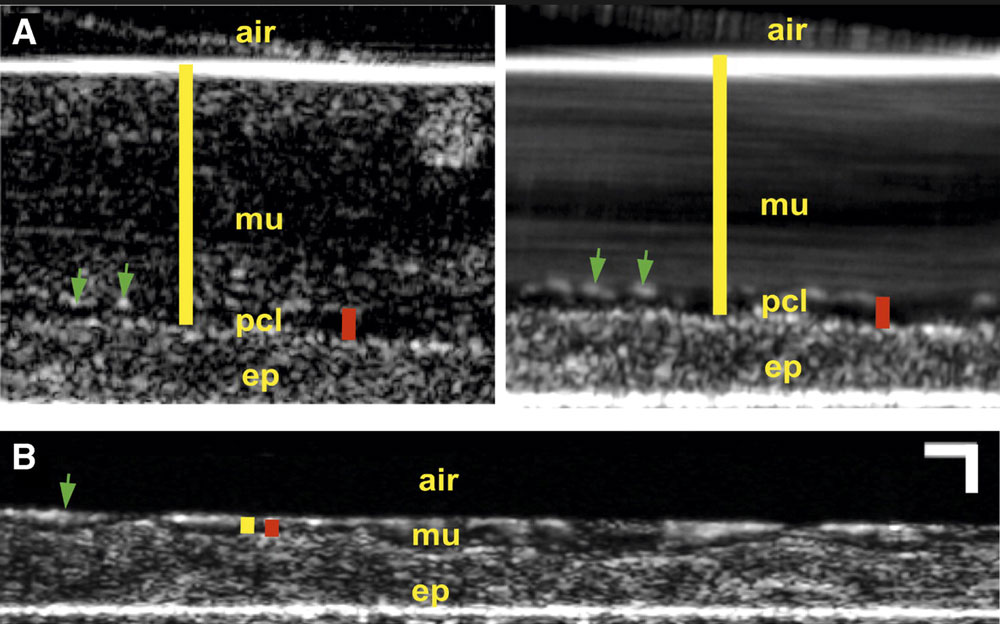 Images from the microOCT system, taken from the 2014 paper "A functional anatomic defect of the cystic fibrosis airway" in the American Journal of Respiratory and Critical Care Medicine. Reprinted with permission of the American Thoracic Society. Copyright © 2018 American Thoracic Society. Cite: Birket, Chu, Liu, Houser, Diephuis, Wilsterman, Dierksen, Mazur, Shastry, Li, Watson, Smith, Schuster, Hanes, Grizzle, Sorscher, Tearney, Rowe/2014/"A functional anatomic defect of the cystic fibrosis airway"/American Journal of Respiratory and Critical Care Medicine/Vol. 190, No. 4/Pages 421-32. The American Journal of Respiratory and Critical Care Medicine is an official journal of the American Thoracic Society.
Images from the microOCT system, taken from the 2014 paper "A functional anatomic defect of the cystic fibrosis airway" in the American Journal of Respiratory and Critical Care Medicine. Reprinted with permission of the American Thoracic Society. Copyright © 2018 American Thoracic Society. Cite: Birket, Chu, Liu, Houser, Diephuis, Wilsterman, Dierksen, Mazur, Shastry, Li, Watson, Smith, Schuster, Hanes, Grizzle, Sorscher, Tearney, Rowe/2014/"A functional anatomic defect of the cystic fibrosis airway"/American Journal of Respiratory and Critical Care Medicine/Vol. 190, No. 4/Pages 421-32. The American Journal of Respiratory and Critical Care Medicine is an official journal of the American Thoracic Society.
In the lab, microOCT “lets us see things no one has ever seen before,” he explains — “features of the pathology of CF that we didn’t imagine, including what’s wrong with CF mucus and what we might need to do to improve it.” That’s especially important with new mucus-focused drugs on the horizon. The team’s latest advance is a miniaturized version of microOCT for patient use, which will let them monitor patient reactions to drugs in near real time, and personalize therapy.
microOCT video of mucus movement in non-cystic fibrosis human bronchial epithelial (HBE) cells (top) and cystic fibrosis HBE cells.
Video supplement from the article "A functional anatomic defect of the cystic fibrosis airway," 2014. Reprinted with permission of the American Thoracic Society. Copyright © 2018 American Thoracic Society. Cite: Birket, Chu, Liu, Houser, Diephuis, Wilsterman, Dierksen, Mazur, Shastry, Li, Watson, Smith, Schuster, Hanes, Grizzle, Sorscher, Tearney, Rowe/2014/"A functional anatomic defect of the cystic fibrosis airway"/American Journal of Respiratory and Critical Care Medicine/Vol. 190, No. 4/Pages 421-32. The American Journal of Respiratory and Critical Care Medicine is an official journal of the American Thoracic Society.
Finding new ways forward
MicroOCT could also help researchers find new ways forward in the treatment of other airway diseases, including COPD and asthma. “The diseased biology of epithelial cells causes almost all the mortality of cystic fibrosis,” says George Solomon, M.D., a researcher in the CF Center and assistant professor in the Division of Pulmonary, Allergy and Critical Medicine. “Damage to the mucus-clearance apparatus is a feature of many other diseases as well. What exactly happens on the cell surface when the CFTR protein is not working, or once it begins to be activated by a drug? This system allows us to start answering these questions. When you can see the cilia and the mucus and the epithelial cells all interacting, in real-time, you have an entirely new perspective.”
The microOCT research began, like much of the CF Center’s work, with a willingness to take risks — and the backing of philanthropic funding, Rowe says. Seed money from the Gwaltney Endowed Chair enabled the project to get started. “With that support, we were able to take this shot on goal,” says Rowe. “That’s always been our philosophy, at that of the CF Foundation — the more shots we can take, the more chances we have to make a difference in the lives of our patients.”
Make a gift to the UAB Cystic Fibrosis Research Center