Case History
Patient is a 66-year-old female with NASH cirrhosis s/p OLT. Specimen: native liver explant. Pictures:
A. Hematoxylin and eosin, 20x. B. Hematoxylin and eosin, 100x. C. PAS w/diastase, 40x
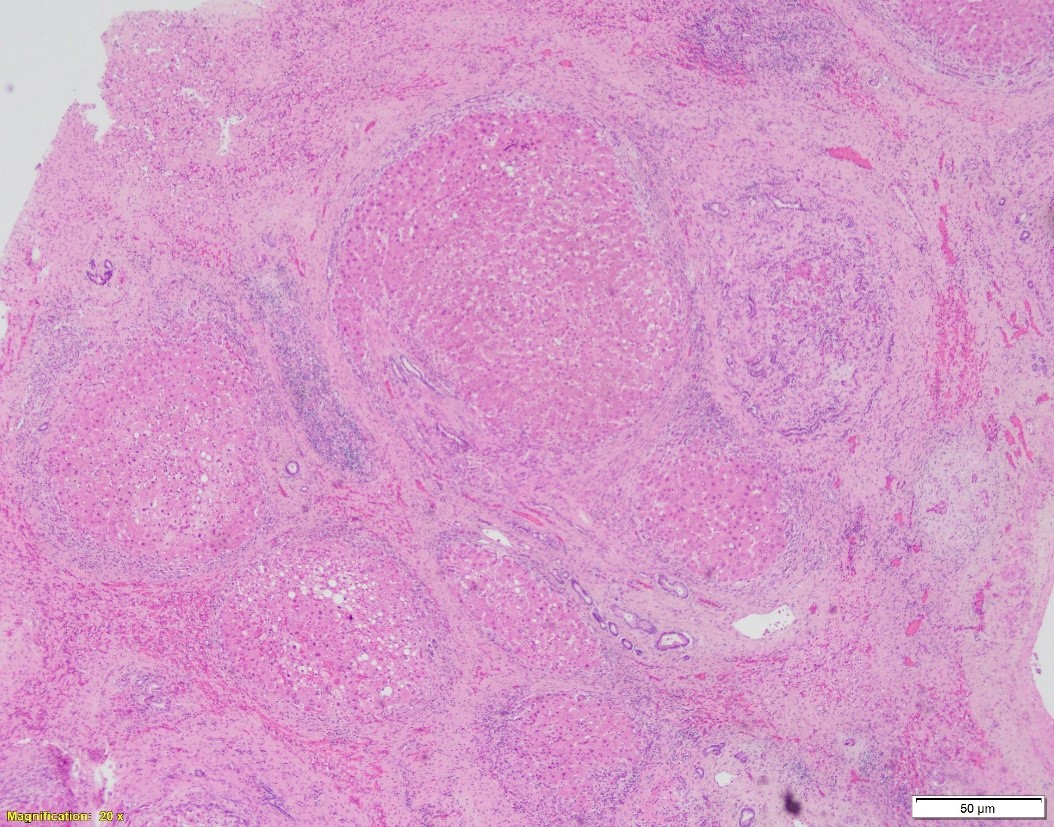
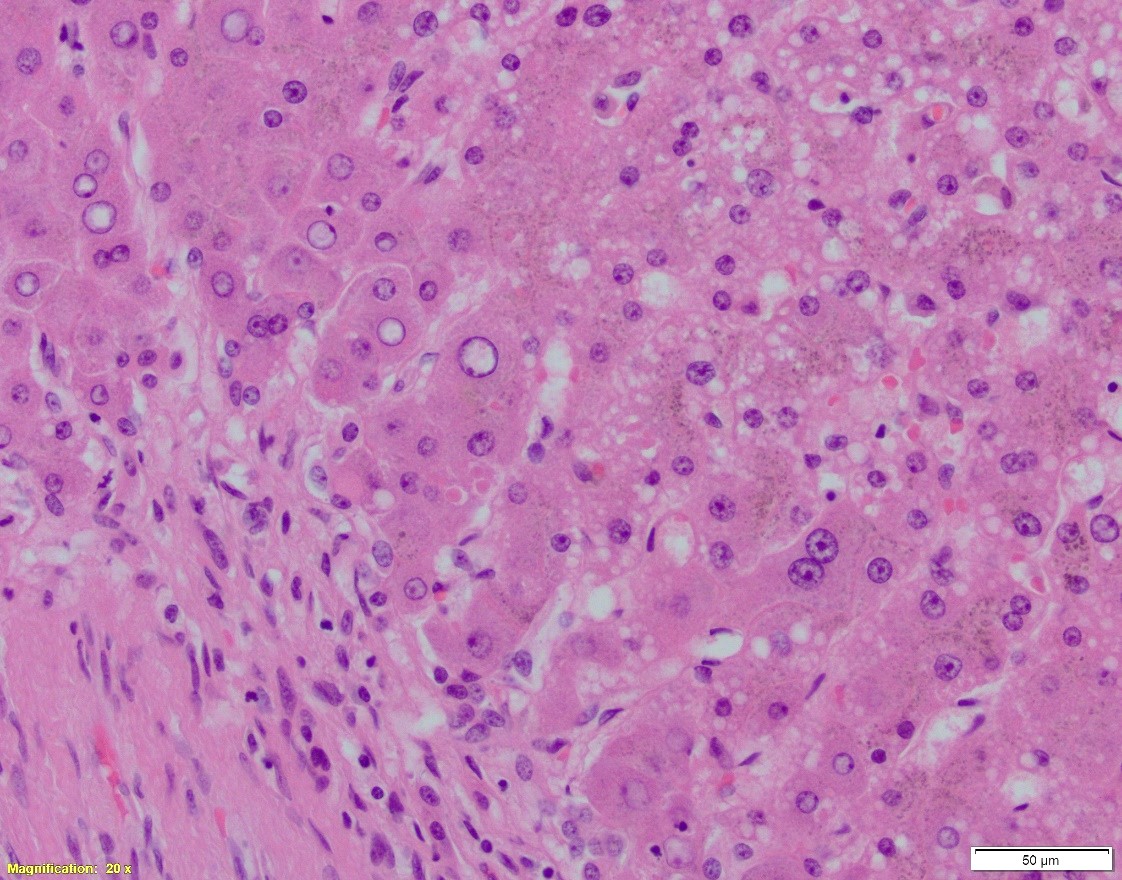
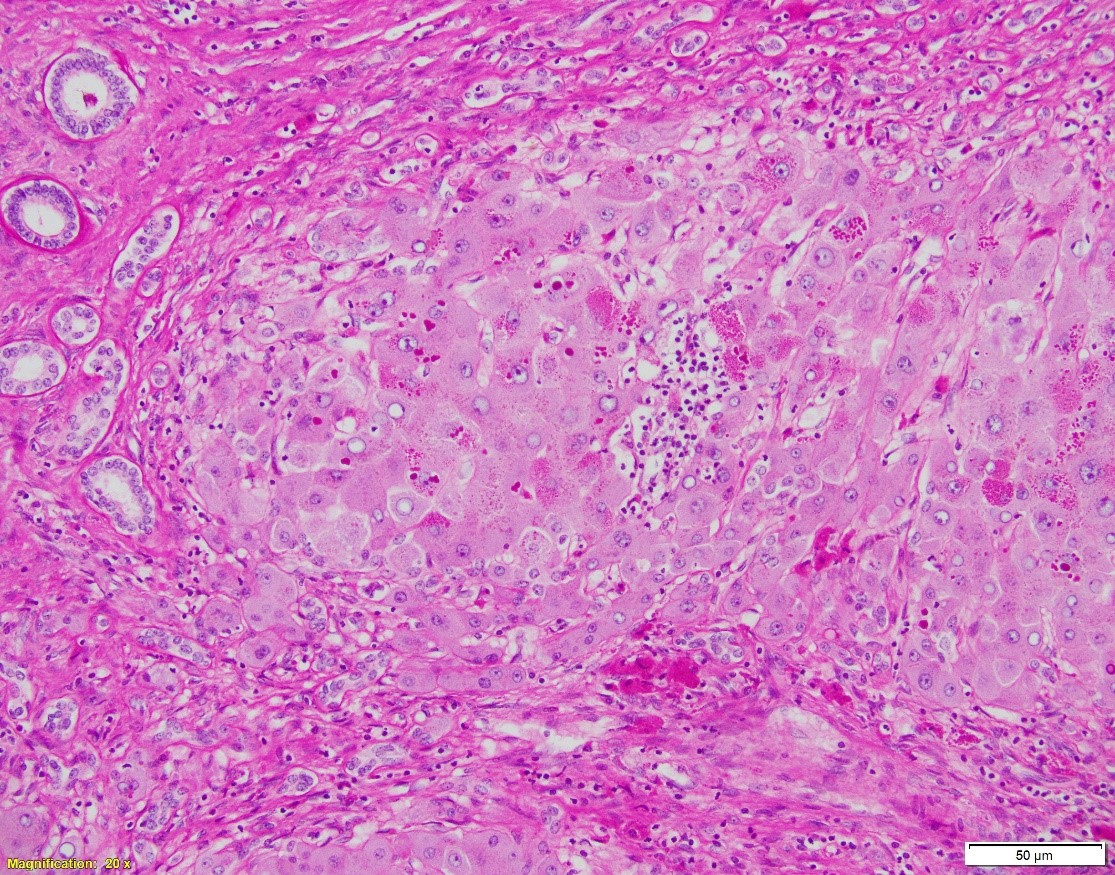
Which of the following is NOT true about the patient’s disease?
- Most common mutations include C282Y and H63D
- The intracytoplasmic globules positive on both PAS w/ and w/o diastase
- The disease can grossly and microscopically present as cirrhosis.
- Ursodeoxycholic acid can improve symptoms, while liver transplantation is utilized for definitive treatment.
Answer: A. the mutations mentioned (C282Y and H63D) are consistent with the diagnosis of Hereditary Hemochromatosis, but not A1-antitrypsin deficiency, see discussion.
Discussion:
A1-antitrypsin deficiency is a genetic disorder caused by defective production of the enzyme by hepatocytes, leading to its accumulation in liver and decrease serum levels. The affected individuals carry mutations in the SERPINA1 gene, with normal phenotype designated as PiM and most common deficiency variants PiS and PiZ. Primary manifestation of disease is in the lungs where it causes emphysema. In children and neonates, the disease can manifest as neonatal hepatitis and jaundice, which later subsides. Adults may develop chronic liver disease, including elevated LFTs, advanced cirrhosis, or hepatocellular carcinoma.
In cirrhotic liver removed for transplantation, A1-antitrypsin deficiency grossly presents as micronodular or mixed micro- and macronodular pattern of cirrhosis. Microscopic findings include intracytoplasmic eosinophilic globules in periportal hepatocytes, which does not always correlate with the clinical liver disease. PAS w/diastase is helpful to distinguish A1-antitrypsin globules from glycogen. In infants, the globules may be absent or hard to detect; in these children, however, the disease may present as liver injury in form of neonatal hepatitis, cholestatic hepatitis, or extrahepatic biliary atresia, with variable degrees of fibrosis. In adults, besides the presence of A1AT globules, the changes may include mild portal inflammation and bile ductular proliferation, mild steatosis, and variable fibrosis.
Other eosinophilic inclusions that can mimic A1AT deficiency on H&E are mega-mitochondria, macrophages containing granules, immunoglobulin and fibrinogen (afibrinogenemia) globules, and rarely, lipofuscin.
Reference: Surgical Pathology of the Liver. Michael Torbenson; Roger Moreira; Lizhi Zhang. 2018 Wolters Kluwer.