Case of the Week
- Details
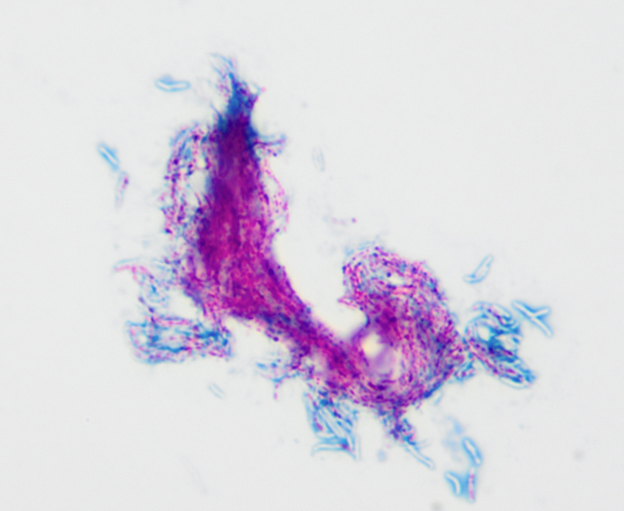
Case History:
Lung nodule FNA from a female with bilateral pulmonary nodules and a history of primary breast neoplasm.
- Details
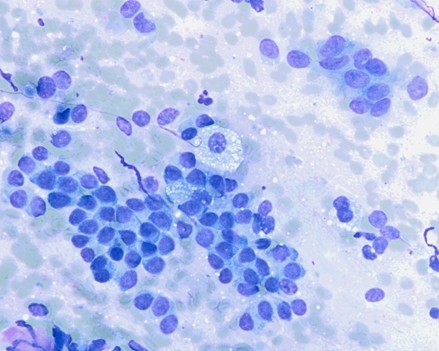
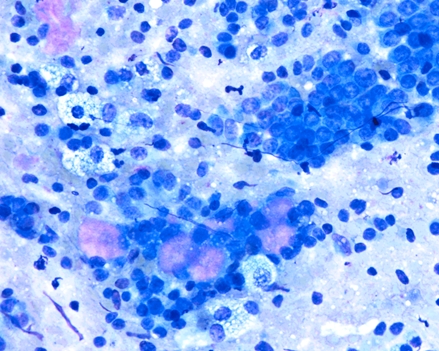
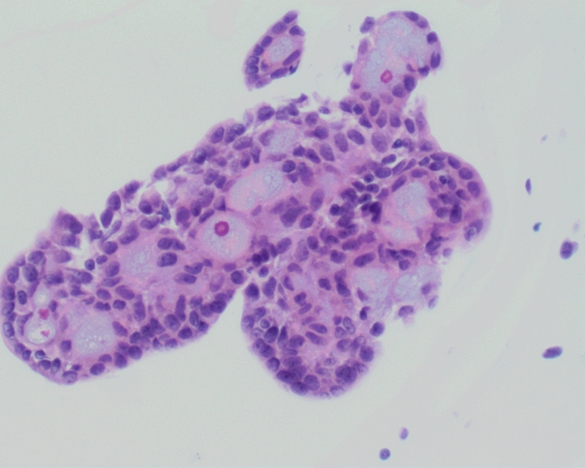
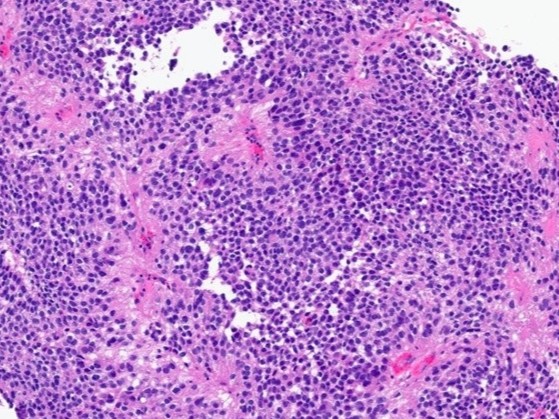
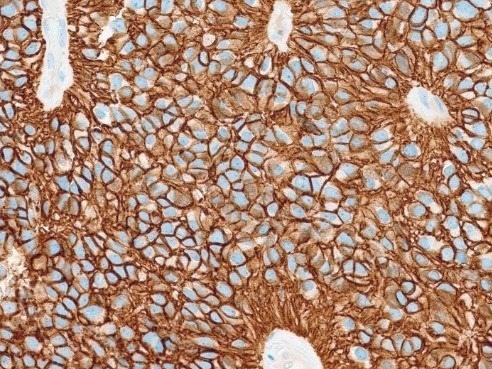
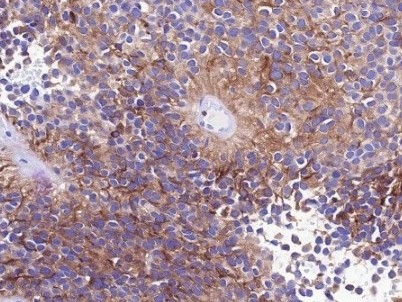
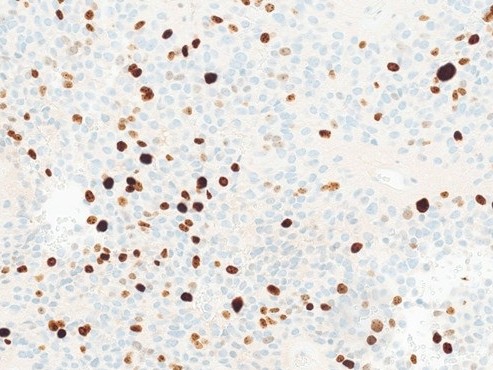
Case History:
A 55 year-old presents with nodular mass in the temporomandibular joint with erosion of the glenoid fossa. Histologic sections from this lesion are shown below.
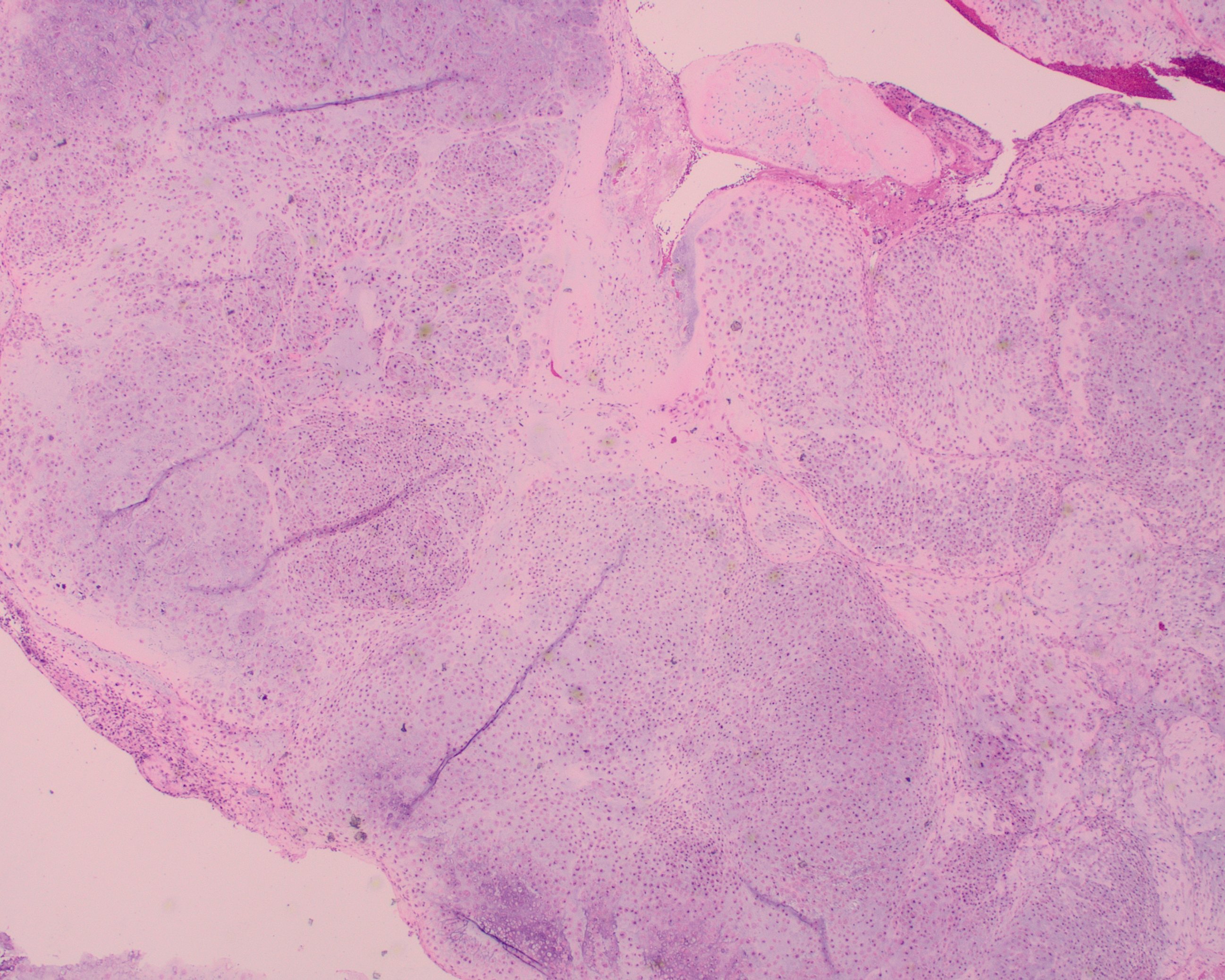
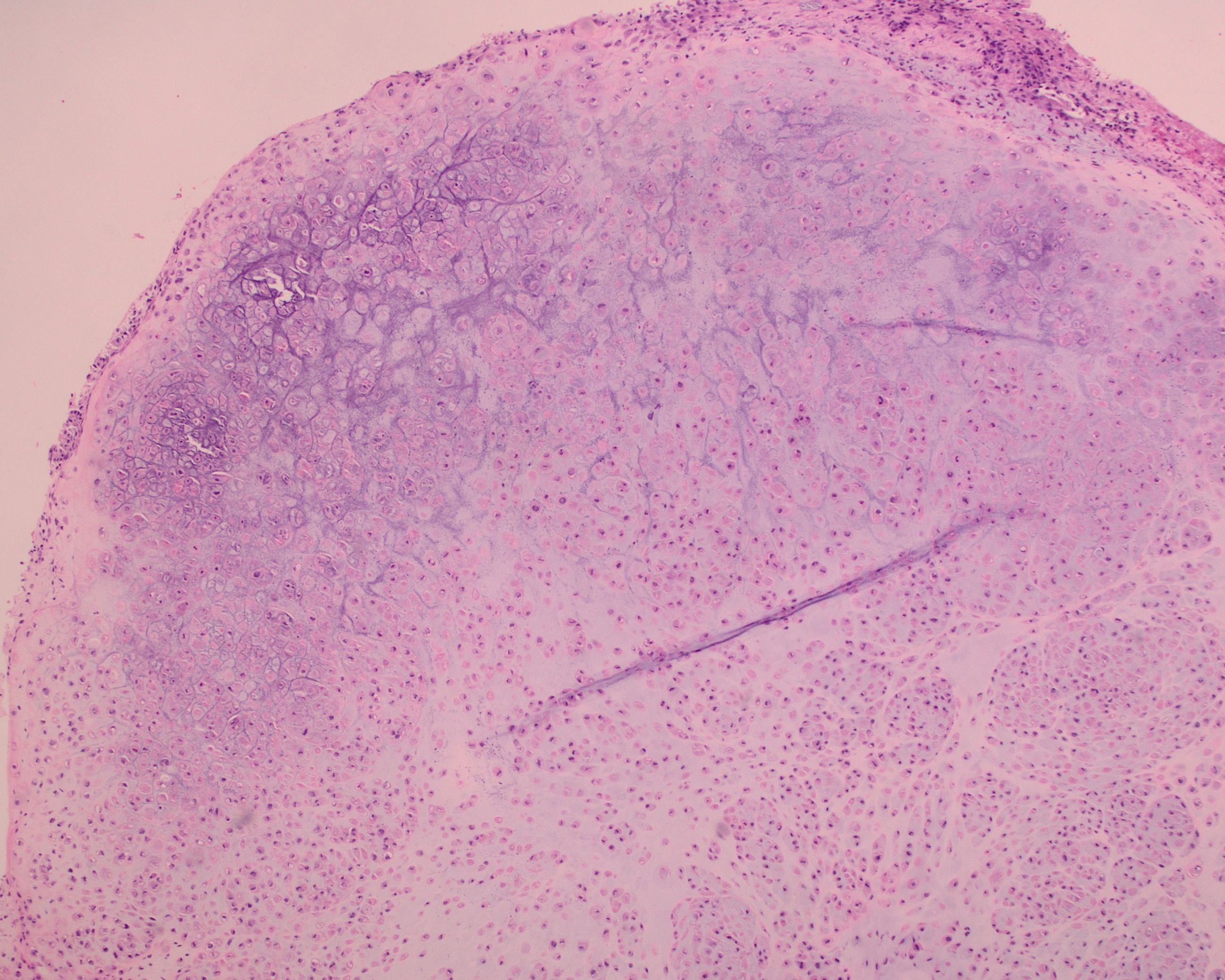
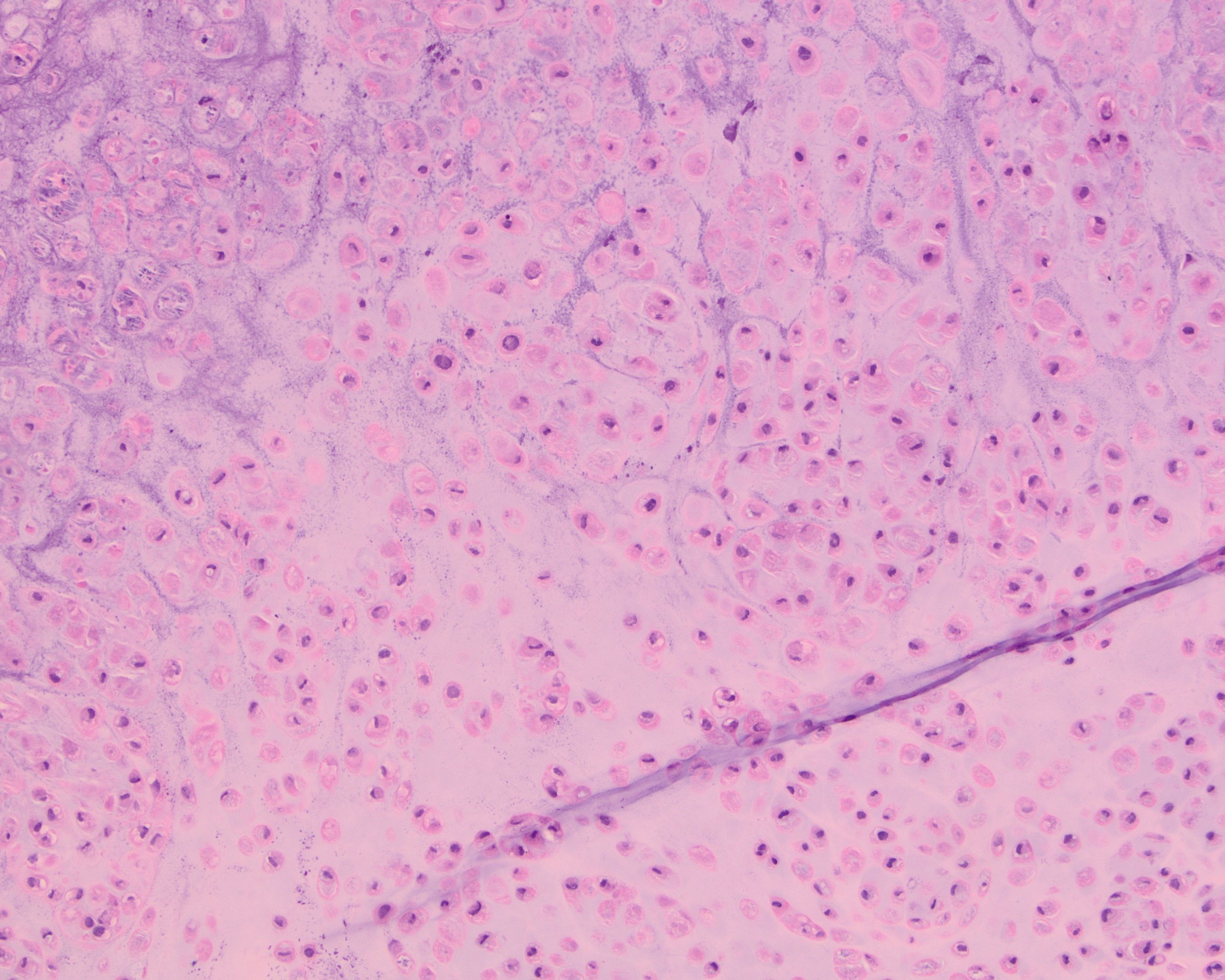
- Details
Case History:
A hospitalized patient with a venous catheter developed fever 15 days after admission to the ICU.
Blood culture sets were collected and the organism shown (Gram stain) was isolated from both anaerobic and aerobic bottles in 2 sets.
Results: Cat=NEG, LAP=POS, and PYR=POS
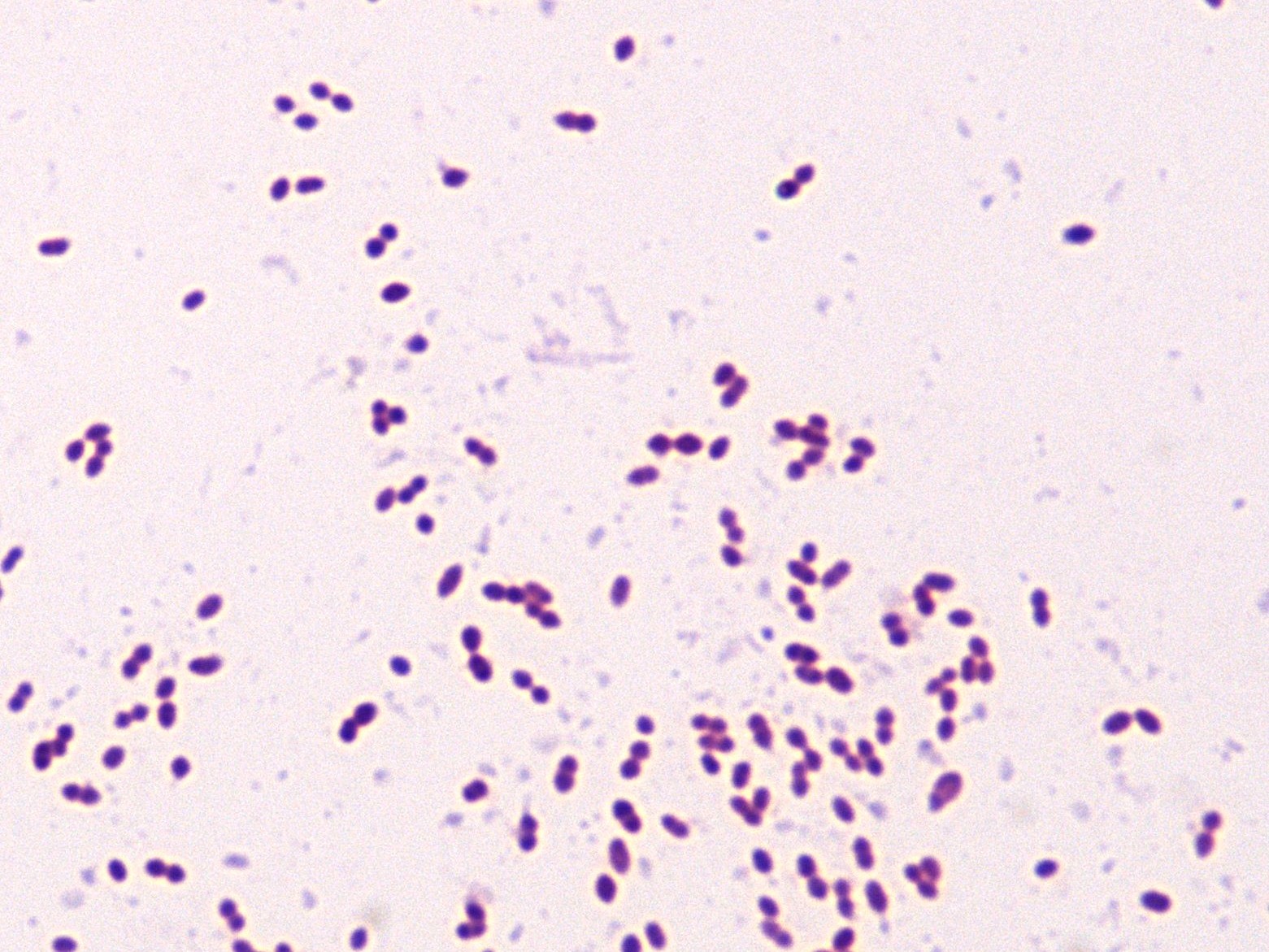
- Details
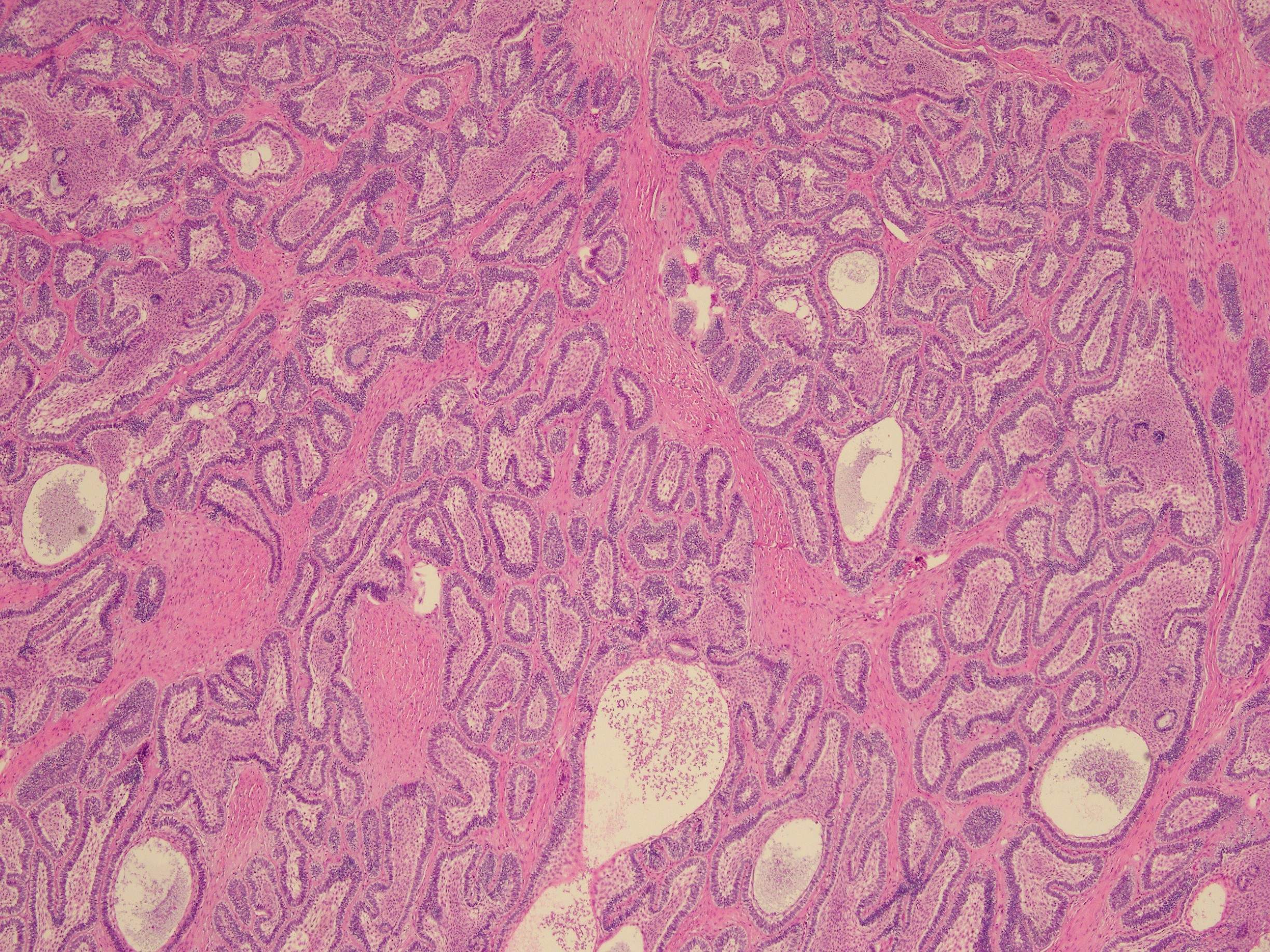
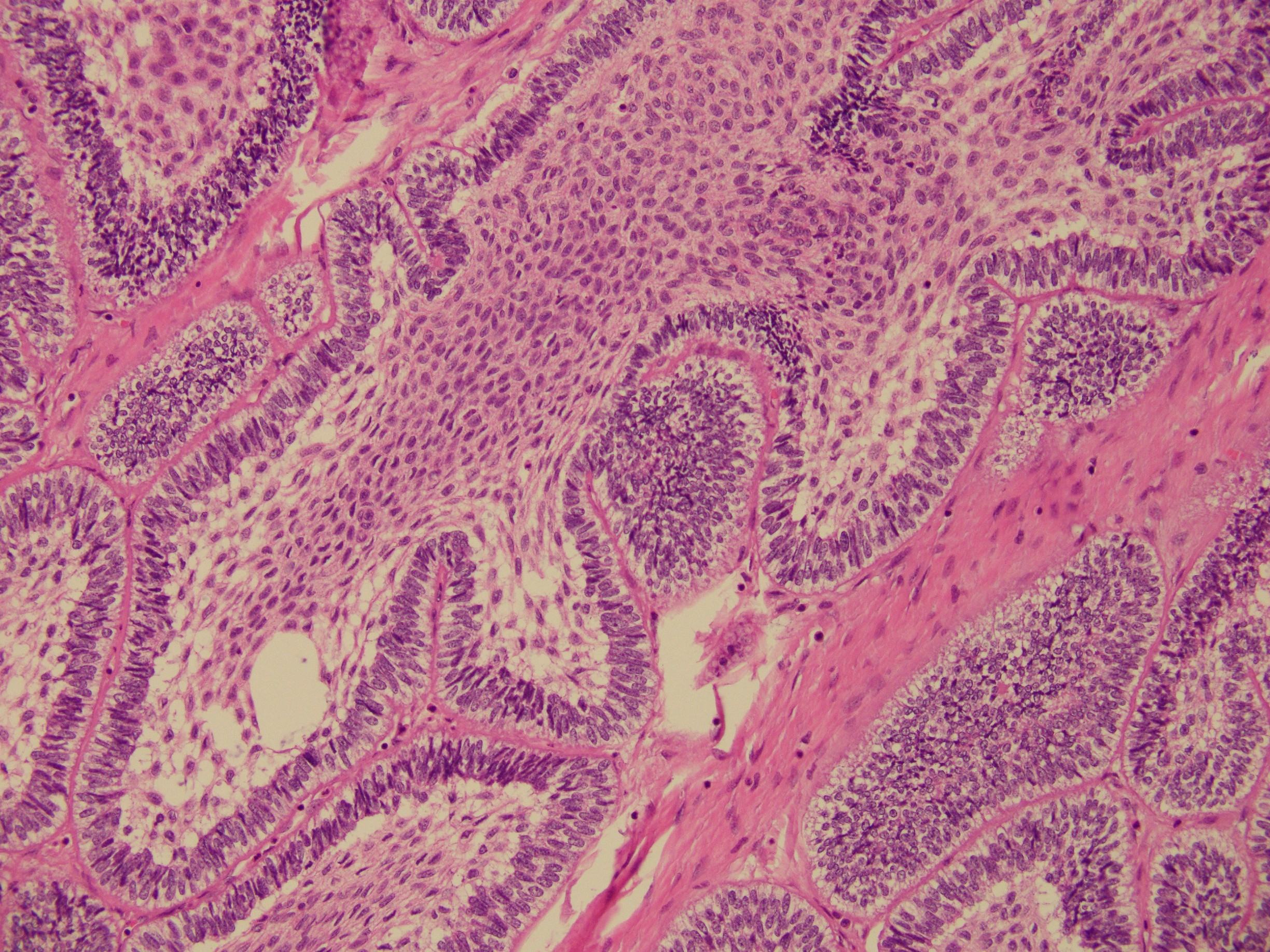
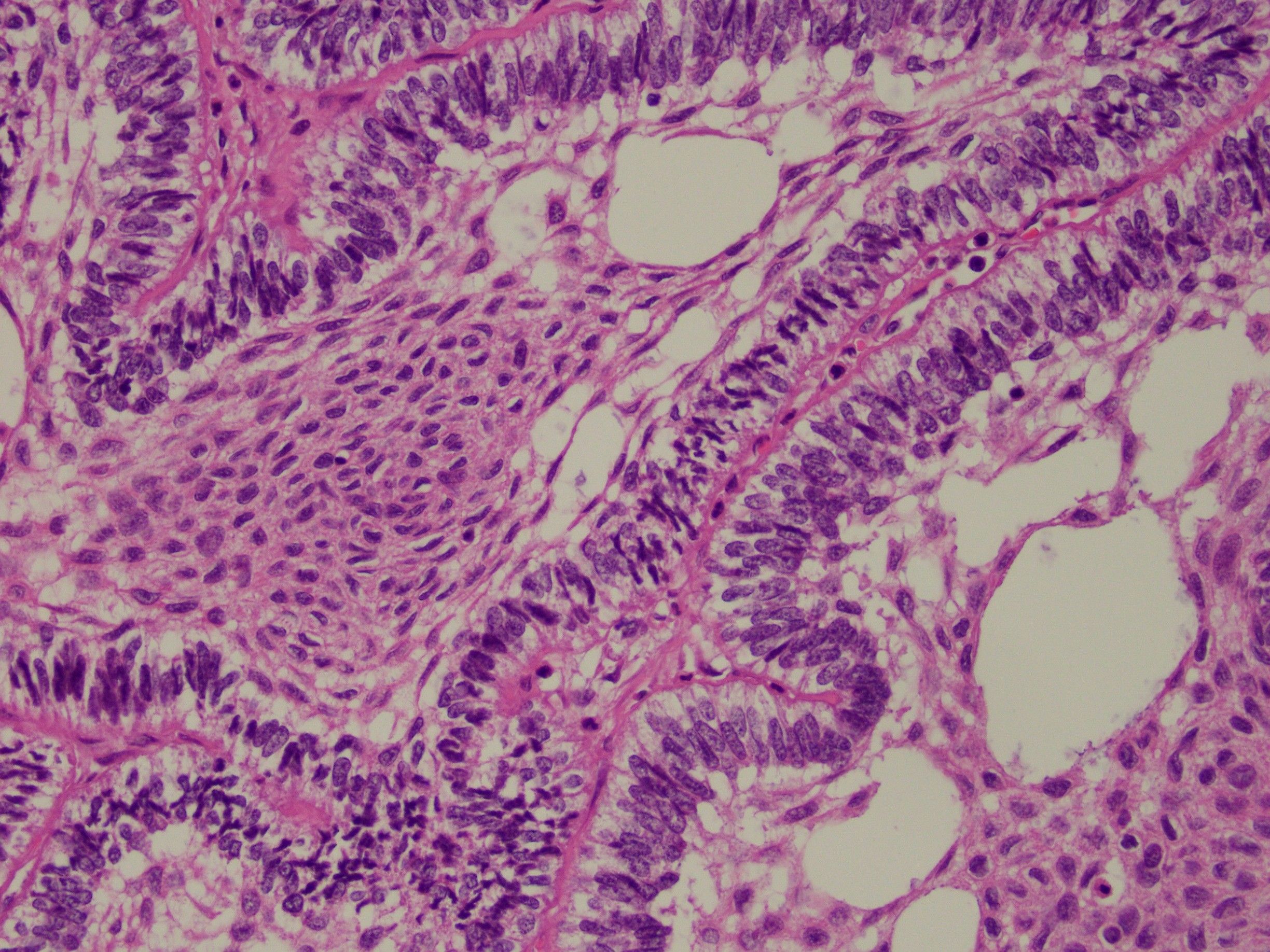
Case History:
A 64 year old female admitted after routine primary care visit revealed leukocytosis (94 x 10^3/cm).
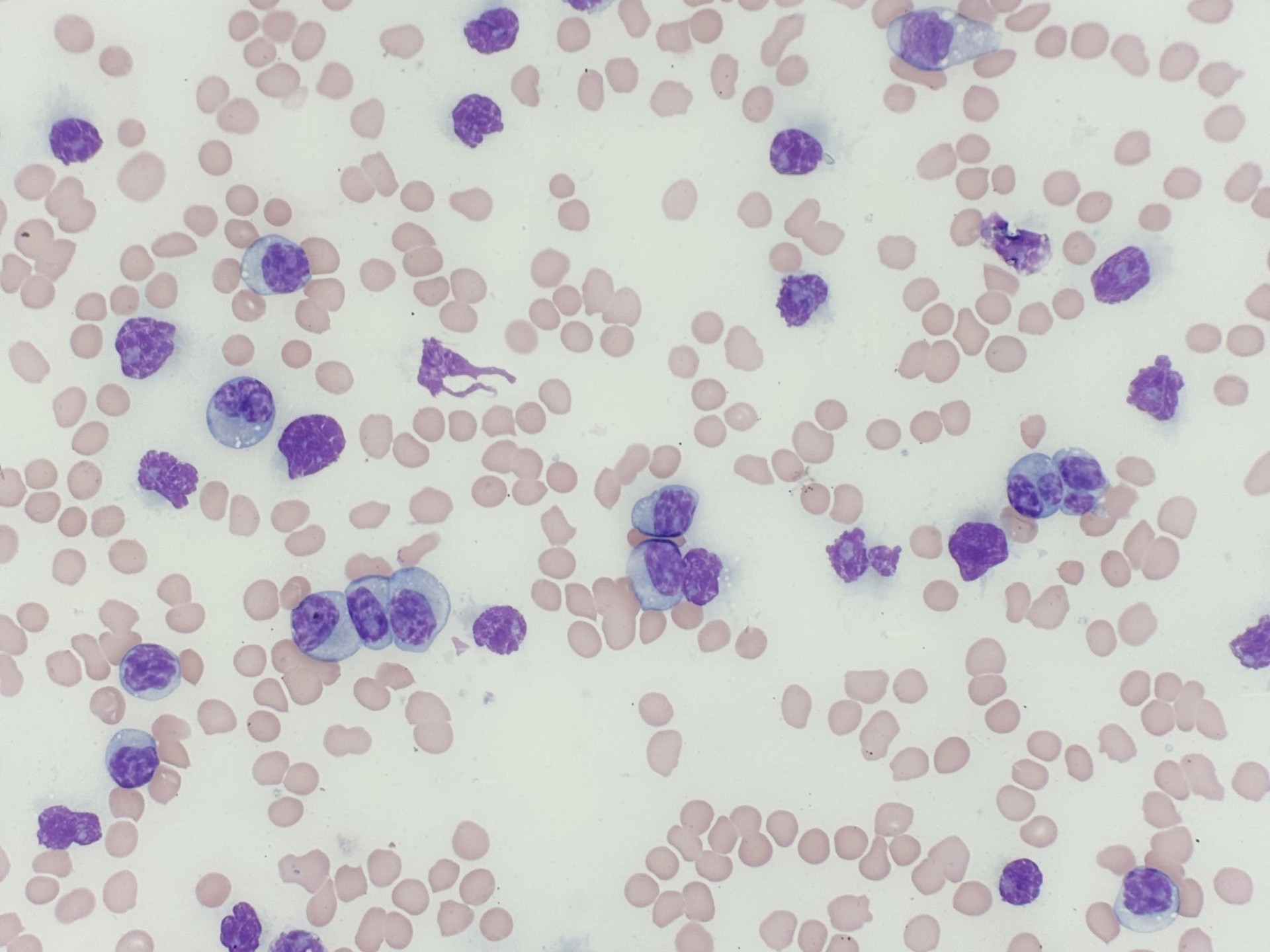
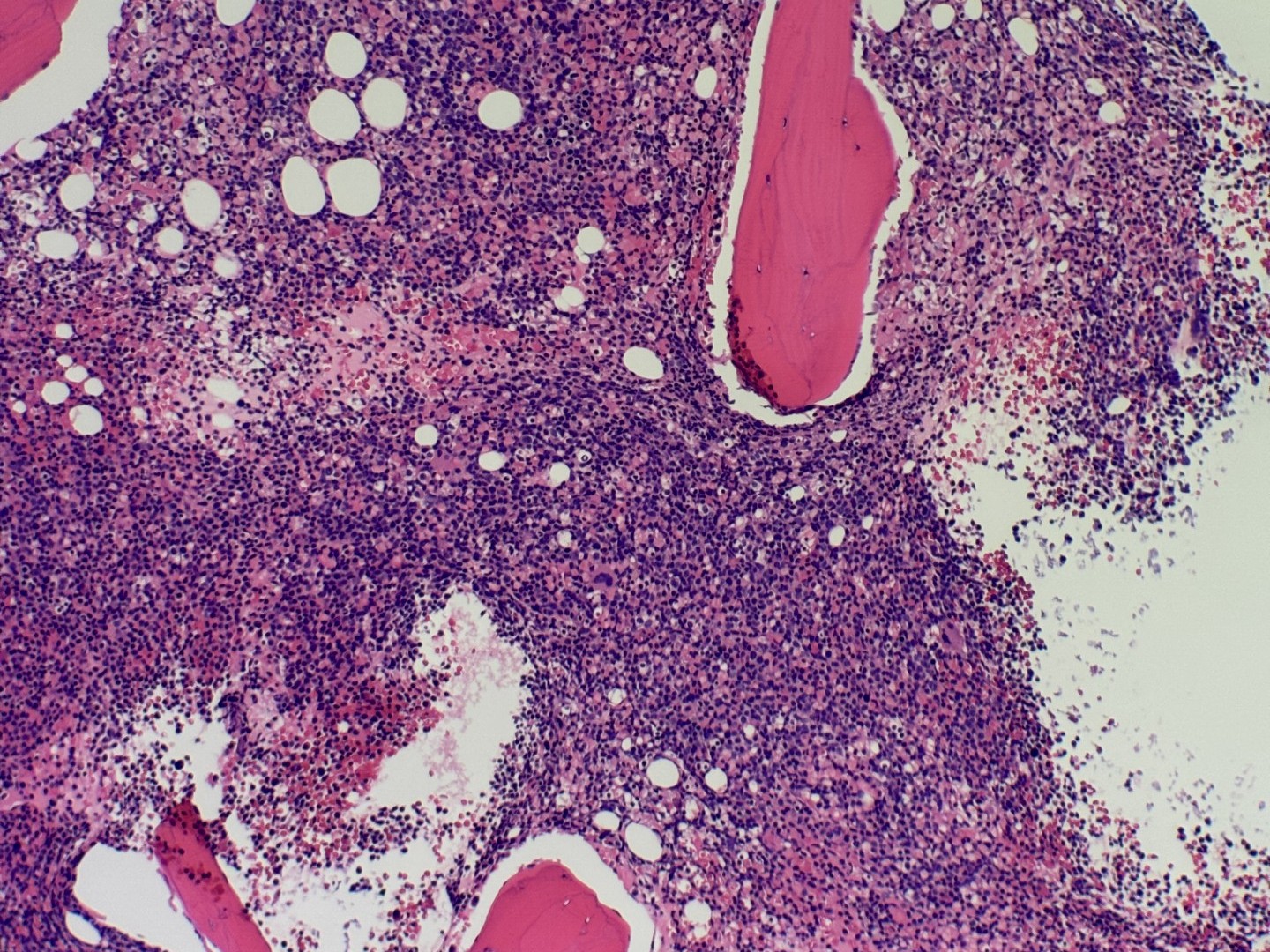
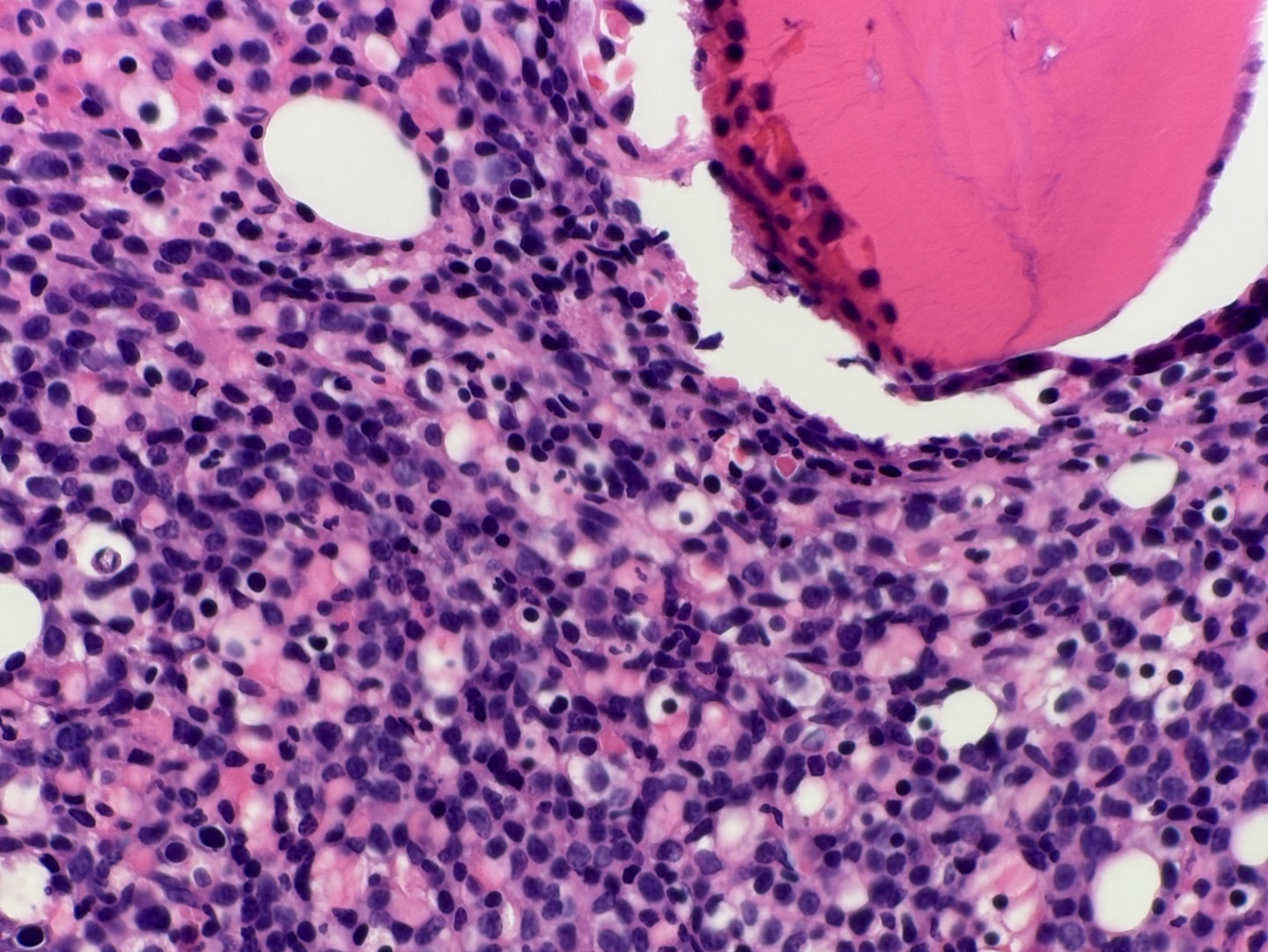

- Details
Case History:
A 55 year old female patient underwent MRI guided biopsy for a non-mass enhancement. She has previously biopsy-proven ipsilateral breast carcinoma.
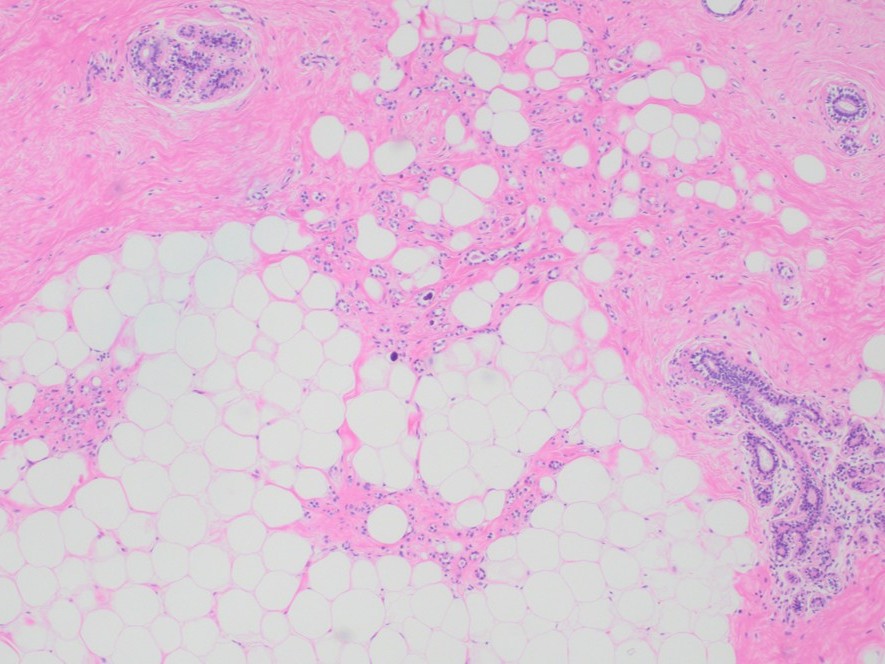
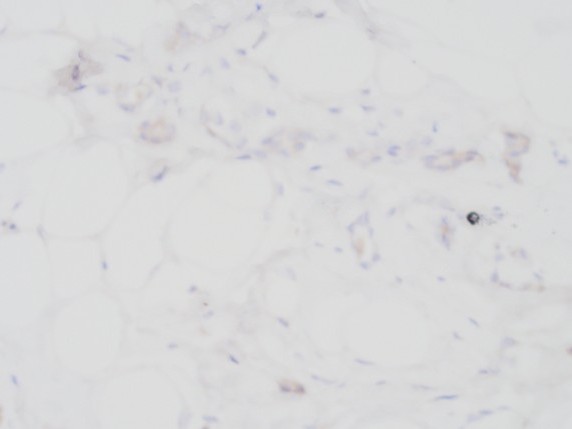
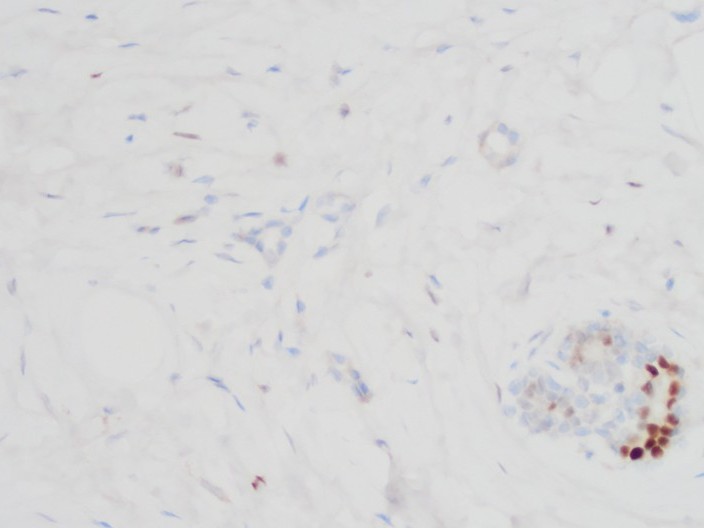
- Details
Case History:
A female patient with cystic fibrosis in her mid-twenties has a positive AFB sputum culture. Below is a Kinyoun stain of positive MGIT tube fluid.