 42 yo F with recently diagnosed multiple myeloma presents with sudden onset of tender erythematous plaques and nodules on the face and extremities
42 yo F with recently diagnosed multiple myeloma presents with sudden onset of tender erythematous plaques and nodules on the face and extremities
What's the underlying condition?
- Granuloma faciale
- Bullous pemphigoid
- Sweet’s syndrome
- Wells’ syndrome
Answer
The answer is “C”, Sweet’s syndrome
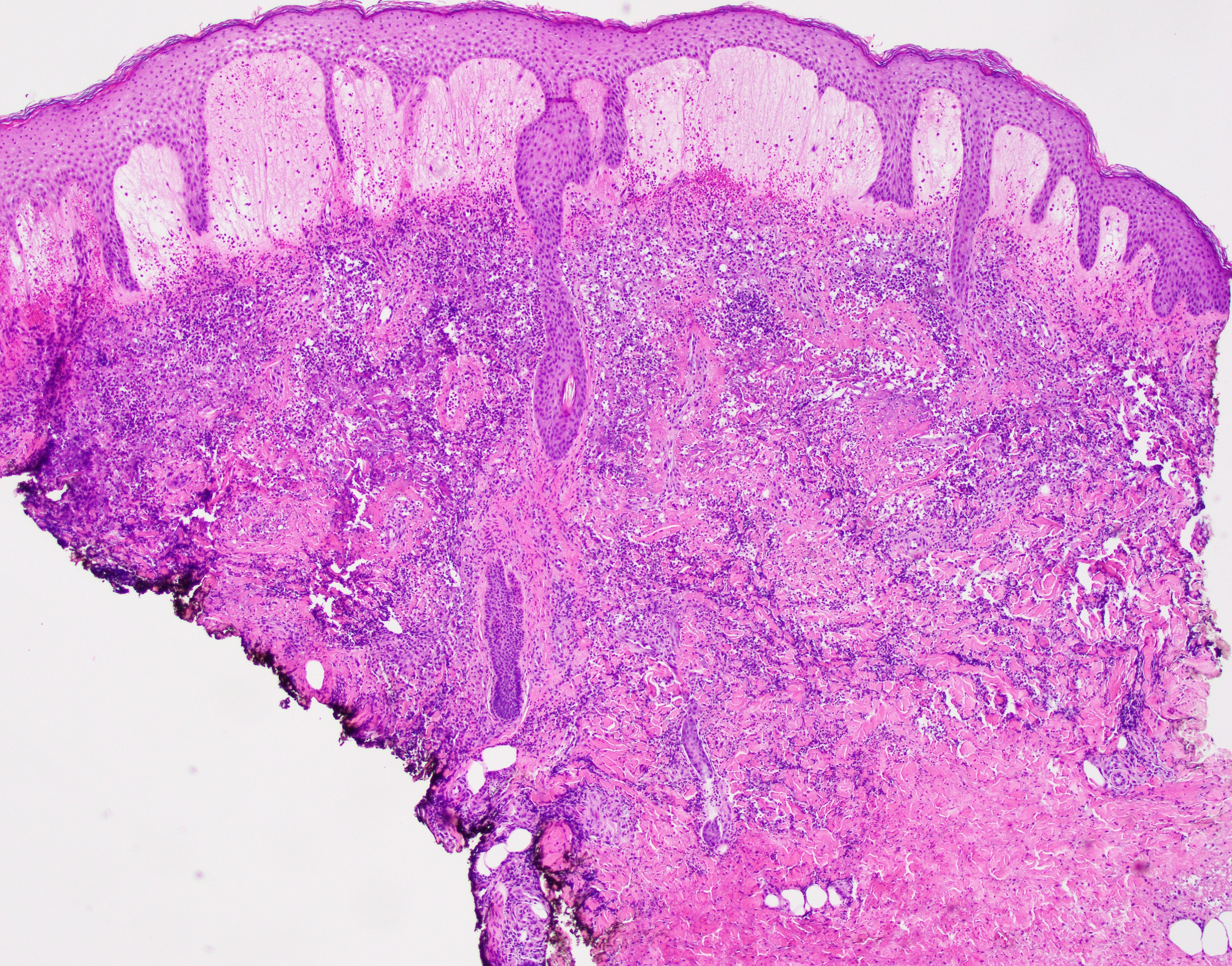
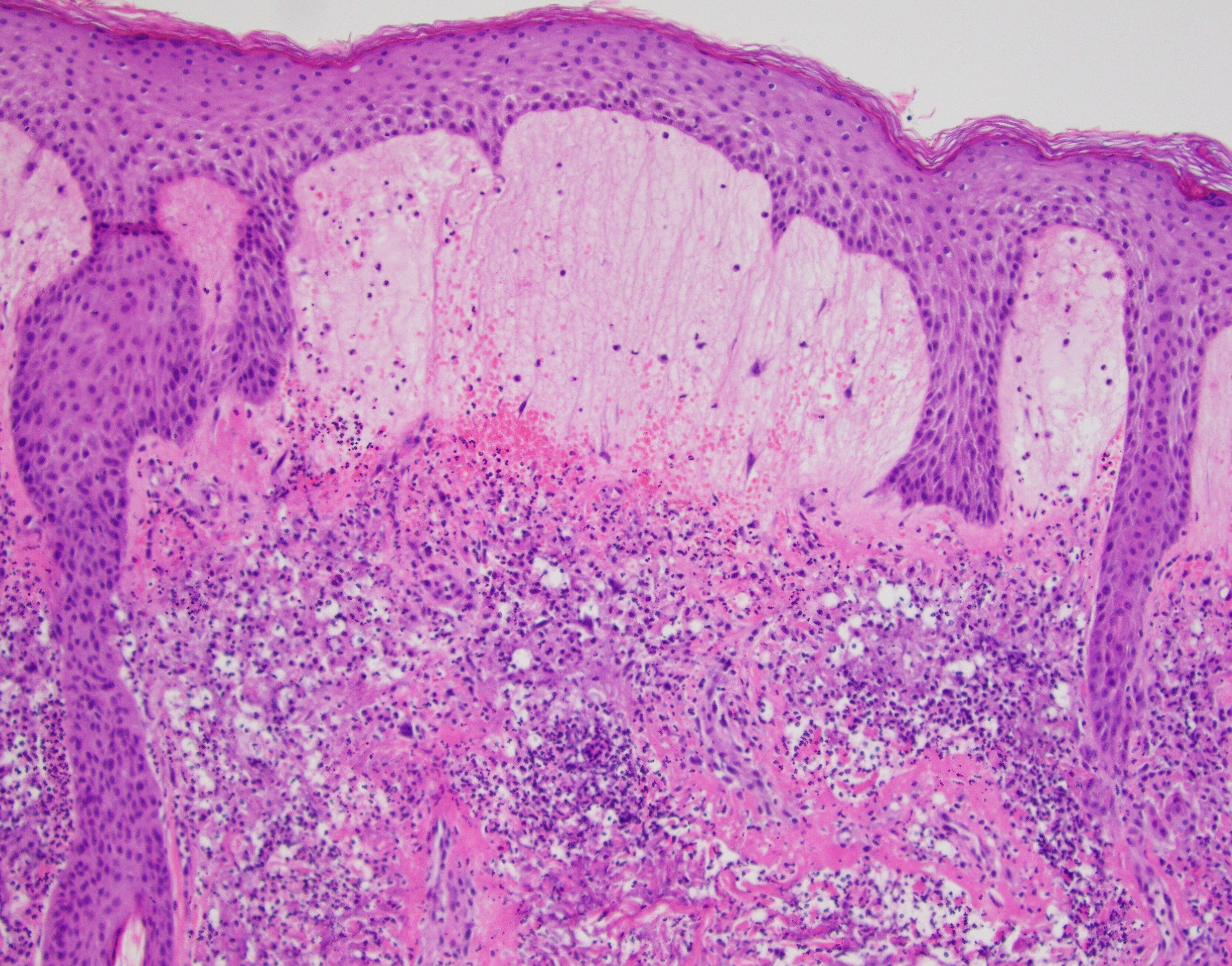
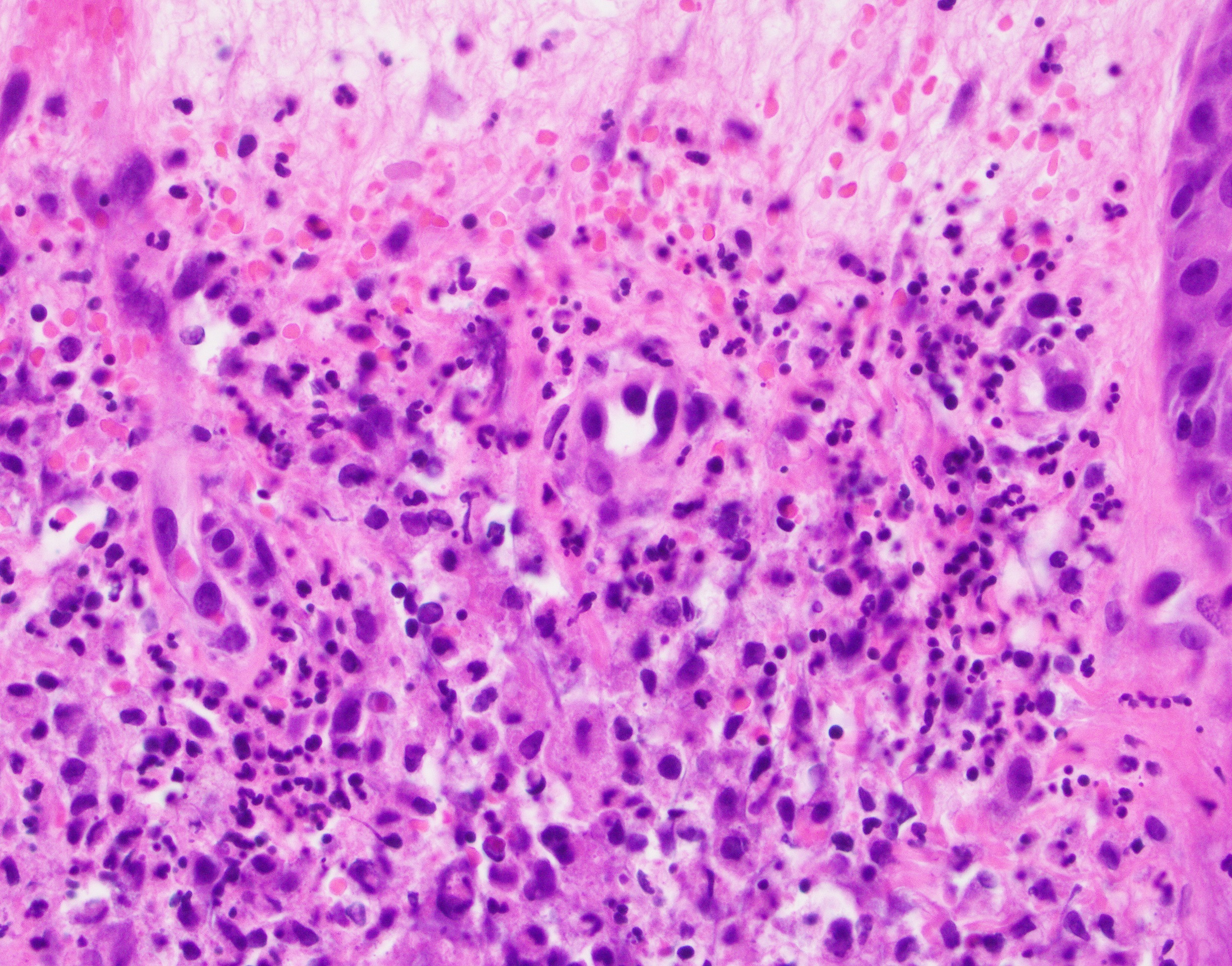
Sweet’s syndrome (SS), named after Dr. Robert Douglas Sweet from Plymouth, England, who first described it in 1964, is a neutrophilic dermatosis of unknown origin, possibly related to an imbalance of granulocyte colony stimulating factors (G- CSF). SS is often related to a variety of hematologic processes, with the most common being acute myelomonocytic type, but it has also been described in association with plasma cell dyscrasia and myeloma. In addition, SS can also be seen in patients with solid malignancies as well as with autoimmune and infectious diseases. Patients with SS often present with dramatic and abrupt onset of tender, erythematous and succulent appearing nodules and plaques involving the face, extremities and, less commonly the trunk. The histologic picture is one of the instant pattern recognition entities in dermatopathology with the characteristic prominent edema of the papillary dermis that overlies a dense, “dirty”-appearing, inflammatory infiltrate predominantly composed of neutrophils. Rare presentations include the so-called “histiocytoid” variant of Sweet’s syndrome, which shows an abundance of immature but non-neoplastic myeloid/monocytic precursors, instead of mature neutrophils.
Granuloma faciale (GF) (option A) is a dermatosis that is restricted to the face (extrafacial forms have been reported) and also shows a dermal “dirty”- appearing inflammatory infiltrate that is somewhat centered around vessels. However, the inflammatory infiltrate in GF is separated from the epidermis by a thin grenz zone and is composed of neutrophils, plasma cells, eosinophils and lymphocytes (“full house”).
Bullous pemphigoid (BP) (option B) is a relatively common subepidermal blistering disease with eosinophils that presents as tense bullae that are preceded by an eczematous/urticarial dermatosis. This disease is related to autoantibodies against the “bullous pemphigoid antigen”, BP180 and BP280, proteins related to the hemidesmosome machinery. The histopathologic changes of classic BP includes the presence of scattered to numerous eosinophils flanking and populating a subepidermal bulla. Classic immunofluorescence findings include linear binding of IgG and/or C3 along the basal membrane zone.
Finally, Wells’ syndrome (WS) (option D) or eosinophilic cellulitis is a dermatosis of unknown etiology that has a somewhat similar clinical presentation to SS but is characterized by development of slate-grey morphea-like induration of the lesions that resolve with no sequelae. WS is usually idiopathic but it can arise in association with infectious and autoimmune processes. Histopathologically, there is a pan dermal and subcutaneous infiltrate composed of eosinophils with the accompanying characteristic “flame figures” that are product of massive eosinophil degranulation of eosinophil granule major basic protein associated with dermal collagen which is surrounded by palisading histiocytes and occasional giant cell.
References
Patterson, James. Weedon's Skin Pathology, 4th Edition, Churchill Livingstone, 2015
Contributed by Carlos Prieto Granada, M.D.