Case History
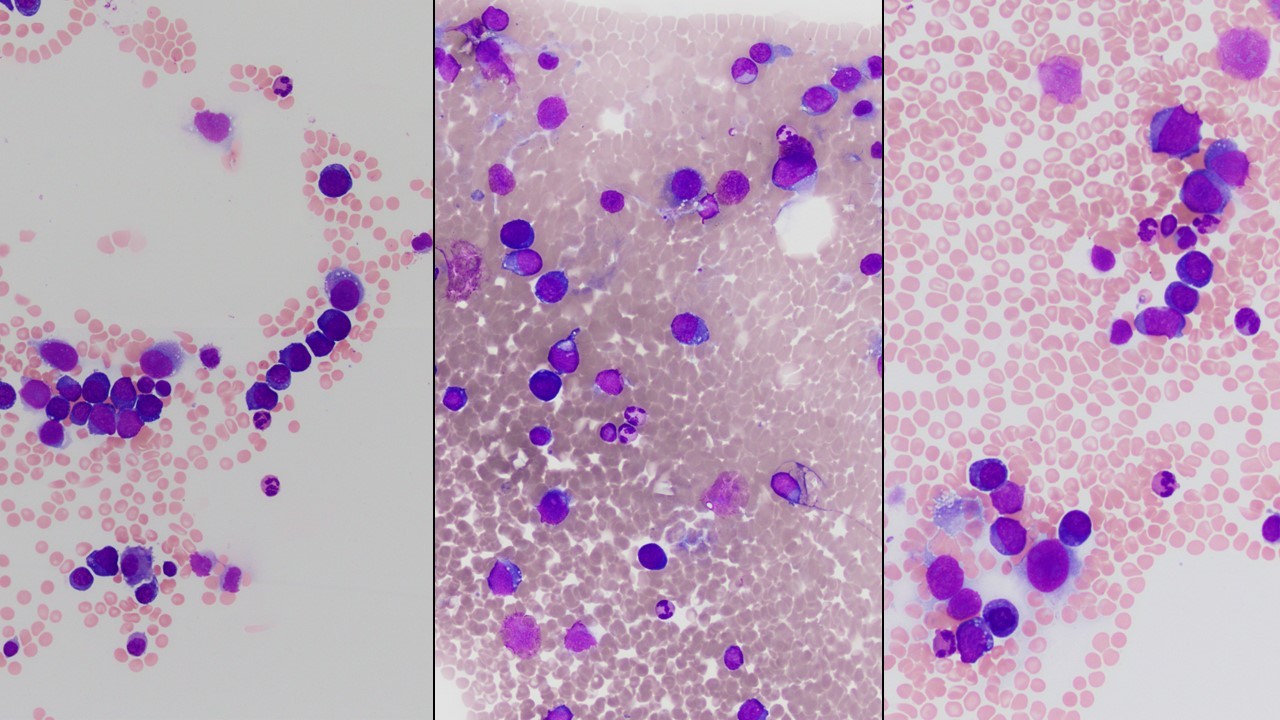
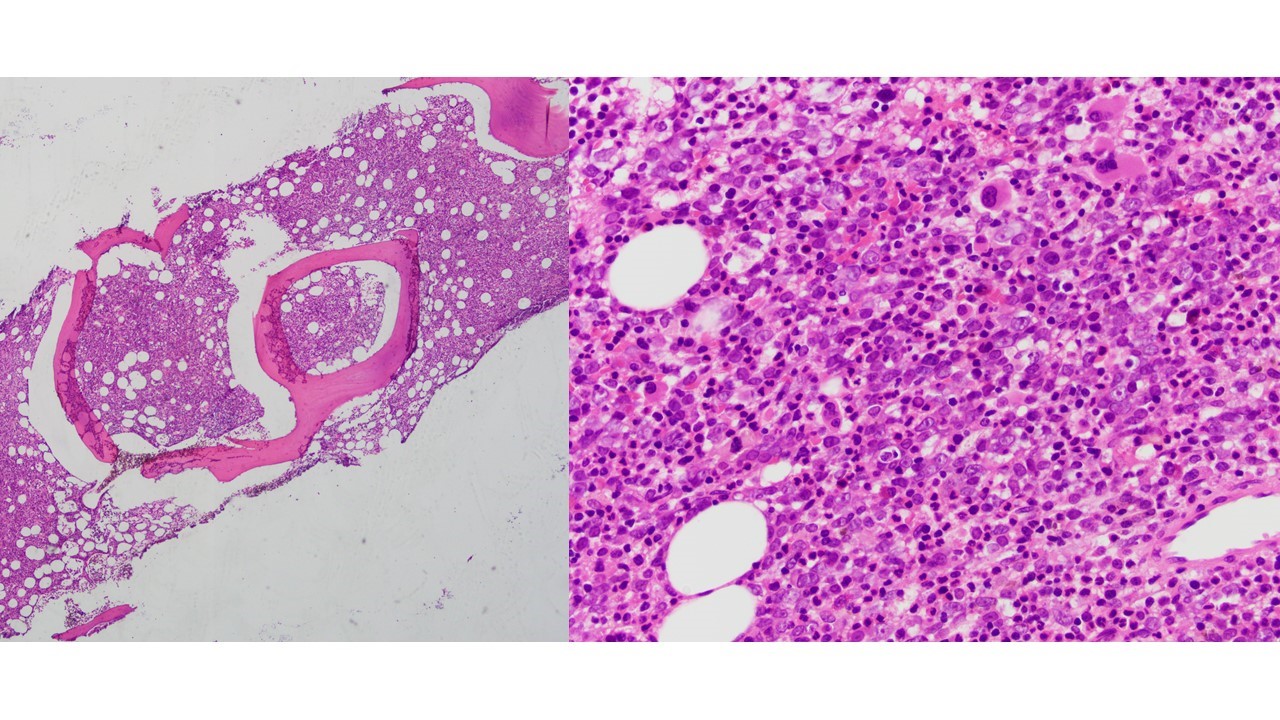
A Pt w/ low plt & BM Bx showing 60% blasts.
Which of the following do not fit with the findings?
- t(1;21)
- inv(3)(q21q26.2)
- t(9;11)
- GATA1 mutation
Answer: C. t(9;11)
Discussion
Options A, B and D are genetic abnormalities seen in AML subtypes with megakaryoblastic differentiation other than acute megakaryoblastic leukemia, NOS (AMKL, M7).
General morphologic features:
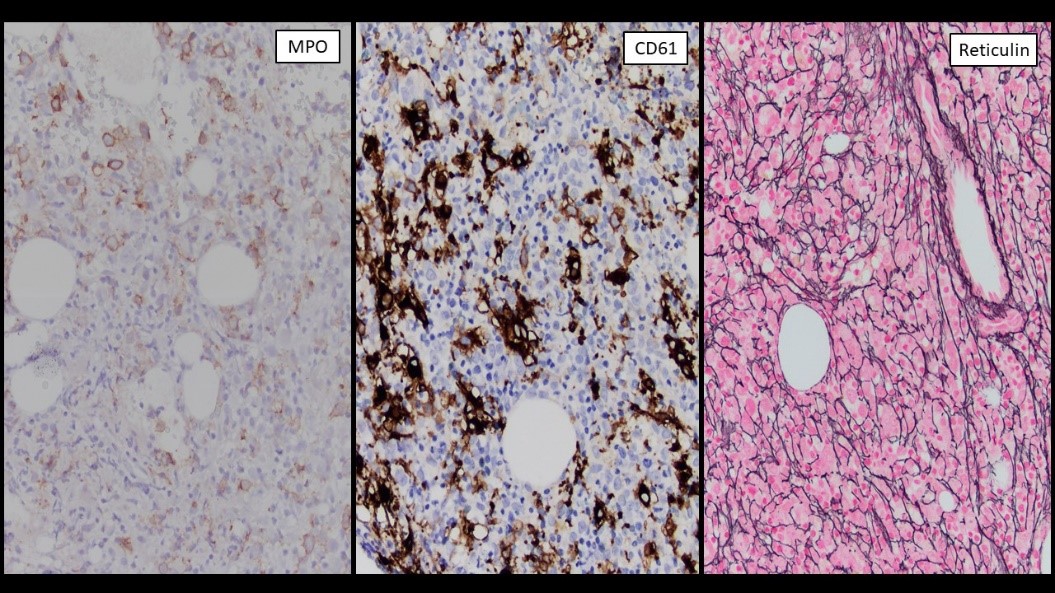
Megakaryoblasts are medium/large cells with dark blue vacuolated agranular cytoplasm. Cytoplasmic projections (blebs and pseudopods) resembling platelets, irregular cytoplasmic borders and cytoplasmic zoning maybe seen. Nuclei are round or slightly indented with finely reticular, dense chromatin and 1-3 nucleoli. Myelofibrosis or increased marrow reticulin is common fibrosis due to megakaryoblast secretion of fibrogenic cytokines, which makes marrow aspiration difficult.
Positive stains:
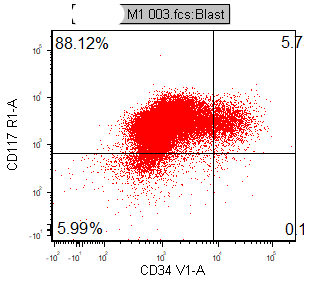
CD41, CD61, CD42b, CD34, CD36, factor VIII and von Willebrand factor
Variable CD13, CD33, CD71, alpha naphthyl acetate esterase, PAS and HLA-DR
Rarely positive for alpha-1-antitrypsin, alpha-1-antichymotrypsin or lysozyme
Negative stains: Myeloperoxidase, Sudan Black B, CD14, CD64 and glycophorin A.
(A) AML with t(1;22)(p13;q13)
Accounts for ≤ 1% of all AML.
Primarily affects infants and young children.
Results in RBM15 and MRTFA (MKL1) fusion gene.
Cases must have ≥ 20% blasts to qualify as AML, even in presence of fusion.
Evaluation for extramedullary disease or myeloid sarcoma should be performed in cases with <20% blasts.
Hepatosplenomegaly &/or bone lesions are common at diagnosis.
Blasts are morphologically and immunophenotypically megakaryoblasts.
Marrow typically shows micromegakaryocytes without multilineage dysplasia.
(B) AML with inv(3)(q21.3;q26.2)
1 - 2% of all forms of AML.
May present as de novo disease or evolve from preexisting myelodysplastic syndrome.
An aggressive form of AML with very poor prognosis and poor response to conventional induction chemotherapy.
More frequent in people younger than 60 years with slight male predominance.
Cases must have ≥ 20% blasts to qualify as AML.
Often clinically characterized by anemia, normal to elevated platelet counts.
Bone marrow features dysplastic small, mono- and bilobed megakaryocytes with accompanying multilineage dysplasia.
Cases < 20% blasts are currently categorized as MDS per 2016 WHO, although this is controversial.
(C) AML with t(9;11); MLLT3-KMT2A
Intermediate-risk disease; more common in children.
20%+ blasts/blast equivalents (monoblasts/monocytes) in peripheral blood or bone marrow, usually myelomonocytic or monocytic (AML M4, M5) and occasionally AML with (M2) or without (M1) maturation.
Monoblasts are large cells with abundant, moderate to intensively basophilic cytoplasm, pseudopods, azurophilic granules, vacuoles; round nuclei, lacy chromatin, and one or more prominent nucleoli.
Promonocytes have basophilic cytoplasm with granules and occasional large azurophilic granules, vacuoles; irregular and delicately convoluted nuclei.
Positive for NSE, CD4 (weak), CD33, CD36 / CD64 coexpression, CD14 (variable), CD45, CD56 and HLA-DR.
Negative for MPO, CD34, and CD117.
(D) Myeloid proliferations related to Down syndrome
Etiology/Pathogenesis: Acquired GATA1 mutations in addition to trisomy 21.
Transient abnormal myelopoiesis (TAM):
Develops in at least 10% of newborns with Down syndrome with average age at diagnosis of 3-7 days.
Hepatosplenomegaly is commonly seen.
Blasts clear from peripheral blood within 3 months after diagnosis, ~5 weeks on average
TAM has also been documented during fetal development (can cause hydrops fetalis).
~66-84% of infants will have spontaneous resolution of their blasts and symptoms without a need for intervention. The remaining infants may need supportive therapy or chemotherapy.
Myeloid leukemia associated with Down syndrome (MLADS):
A spectrum of myelodysplastic syndrome and its evolution into leukemia in children with Down syndrome.
Similar to TAM, trisomy 21 and a somatic GATA1 mutation are thought to be required for the development of ML-DS. However, for this progression, additional genetic and/or epigenetic events are required.
Usually presents between 1-4 years of age.
Can be treated with reduced intensity chemotherapy protocols without stem cell transplantation.
Unlike non-Down syndrome myeloid leukemia, ML-DS has a better prognosis, with better induction remission rates and reduced relapse rates.