 |
Gorgas Case 2019-06 |
|
The following patient was seen in the inpatient department of pediatrics at Cuzco’s Regional Hospital. We would like to thank Dr. Felix Hidalgo, Director of the Hospital for his contribution in presenting this case.
|
 History: 12-yo female presenting with a 3-month history of malaise, fever, diarrhea and generalized lymphadenopathy. Three months before admission, the patient developed loss of appetite and nausea, followed by 3 to 4 daily episodes of watery diarrhea and diffuse abdominal pain. During this period, the patient’s mother noted the presence of several cervical nodules and persistent fever. Symptoms progressed to include bloody diarrhea and a 10 kg weight loss. She was then referred to the Regional Hospital.
Epidemiology: Born and raised in Kiteni, Concepcion (jungle of Cuzco region). She is a student. No recent travel history. No known TB contacts. Two years earlier she had been admitted to our hospital due to a similar clinical presentation.
Physical examination: T 37.7C; HR 90; RR 22; Sat O2 94 %
Pallor +++. Multiple enlarged lymph nodes ranging from 0.5 to 2 cm in diameter in cervical, retroauricular, submandibular, axillary, inguinal and umbilical regions. Chest: clear to auscultation Abdomen is distended. The liver is 2 cm below the costal margin, the spleen is felt crossing the midline. Rest of the physical examination is unremarkable. Laboratory result at admission: Hb 7.3 g/dl; WBC 7020 (N 72% L 12% M 4% E 13%); platelets: 166,000; glucose: 70 mg/dl; albumin: 3.2 g/dl; total bilirubin 1.5 mg/dl; alkaline phosphatase 221; ALT 7 IU/l, creatinine 0.62 mg/dl. HIV, Syphilis, HBsAg: negative. HTLV-1 pending. Stool culture for bacteria and tool examination for ova and parasites: negative
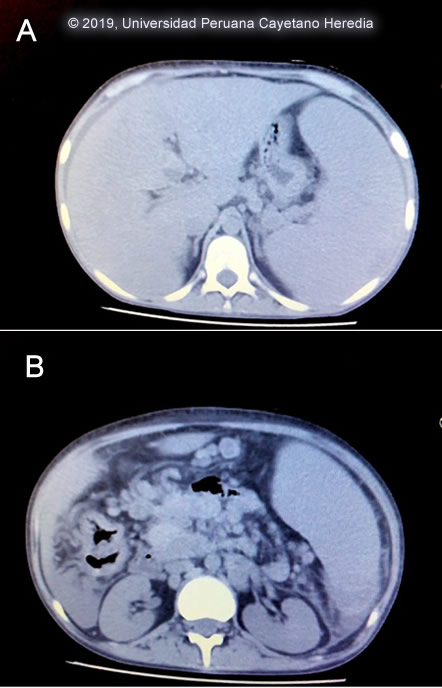
Chest x-ray: no pulmonary infiltrates, no pleural effusion. Abdominal CT-scan [Image A,B]. UPCH Case Editors: Carlos Seas, Course Director / Carlos McFarlane, Associate Coordinator UAB Case Editors: German Henostroza, Course Director / David O. Freedman, Course Director Emeritus |
Diagnosis: Paracoccidioidomycosis, juvenile form
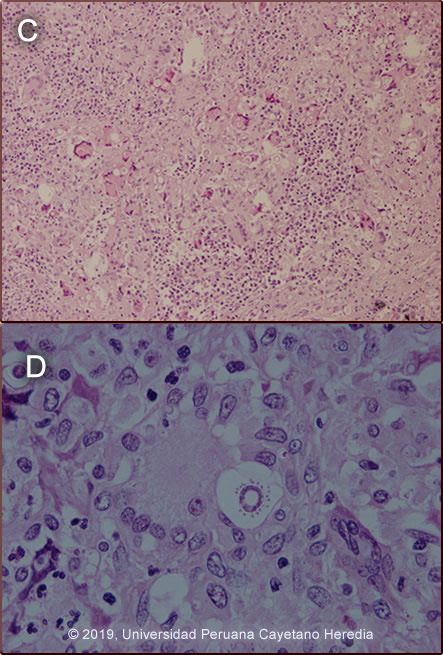
 Discussion: Cervical lymph node biopsy showed that the normal architecture of the lymph node was distorted. The cortex was infiltrated by granulomatous lesions composed of epithelioid cells and multinucleated giant cells containing phagocytosed spherical structures of variable size (5-30um) [Image C], some of them show multiple peripheral budding mimicking a “pilot wheel” suggestive of yeast cells of Paracoccidioides braziliensis [Image D]. If skin lesion were present direct scrapings or aspirates will be positive in the vast majority of cases. Official reading of the CT scan disclosed hepatomegaly with no focal lesions, splenomegaly with no focal lesions diffuse intra-abdominal lymphadenopathy, and no other mass lesions. Records from prior admission 2 years earlier disclosed a similar episode of malaise, fever and subcutaneous nodules. During this episode, she was diagnosed with juvenile paracoccidioidomycosis (PCM). She was treated with amphotericin B and upon discharge was switched to oral itraconazole. Due to economic constraints the family was not able to complete the therapy and stopped it after 5 months without communicating with medical officers. Paracoccidioidomycosis, previously known as South American Blastomycosis, is a systemic fungal disorder endemic in rural areas of Latin America that is acquired through the respiratory route with the lungs constituting the primary target while all other organs (lymph nodes, mucous membranes, skin, adrenals, bone, other) represent secondary manifestations. PCM simultaneously involves more than one organ or system and, as such, should be considered a systemic disorder. PCM is most frequently reported in Brazil, Venezuela, Colombia, Peru and Argentina. It is caused by thermally dimorphic fungi nested within the genus Paracoccidioides (Curr Fungal Infect Rep (2012) 6: 303, Curr Fungal Infect Rep. 2012 Mar; 6(1): 23–34) Recently, molecular tools allowed to revise the taxonomy of this genus determining two species, P. brasiliensis and P. lutzii. Moreover, genetic analysis revealed that P. brasiliensis is not a single species but rather a species complex comprising at least four genetic entities: S1, PS2, PS3, and PS4. However, the clinical impact of this genotypic diversity, if any, has not yet been investigated. (Future Microbiol. 2013 Sep;8(9):1177-91) The infection is usually acquired in the first two decades of life, and it is commonly subclinical, although the fungus can also cause chronic and severe diseases. Most often it is an indolent and relapsing disease causing chronic pulmonary and mucosal manifestations. It is estimated that 10 million people are infected with Paracoccidioides spp. in Latin America, of whom only about 1–2% will develop PCM.(Med Mycol. 2011 Nov;49(8):785-98) PCM predominantly afflicts adult males involved in agriculture and it has a particular gender distribution with a predilection for adult males at a ratio of ≥11 to 1. The disease has mostly a chronic profile with acute/subacute forms accounting for less than 15% of all reports. PCM is uncommon in children and adolescents and the acute/subacute clinical form as seen here is commonest in children and adolescents and represents only 3–5% of all cases. Despite the low incidence, potentially life-threatening and mortality rates may be high. (PLoS Negl Trop Dis. 2017 Mar 29;11(3):e0005500) The chronic form (adult type) of the disease is believed to represent reactivation of latent infection initially acquired via inhalation. The chronic forms represent approximately 94% of all cases in the experience at our institute (100 patients), and approximately 85% in the Brazilian series (Rev Soc Bras Med Trop. 2003 Jul-Aug;36(4):455-9). See examples of this in Gorgas Cases 2005-12, 2009-06, and 2004-05]. We have also previously shown a case of the chronic progressive form of the disease [Gorgas Case 2003-07]. After an initial inhalation event (Clin Infect Dis. 2005 Jan 1;40(1):148-57) which is usually self-resolving, the acute juvenile type systemic disease of PCM may progress from the primary focus of infection without a latency period. Once the disease is established, the patient may develop an acute or sub-acute pattern of clinical manifestations and a rapid clinical deterioration. In a few months or weeks, an acute and febrile rapid course with weight loss and multi-organ compromise is usually observed. As a consequence of the tropism of Paracoccidioides to the monocyte-phagocyte system, a marked involvement of the reticuloendothelial system characterizes this clinical form, (Med Mycol. 2013 Apr;51(3):313-8) and diffuse lymphadenopathy, hepatosplenomegaly, and papular skin lesions with a large burden of yeast forms are usual clinical manifestations as in this patient. Anemia, hypergammaglobulinemia, and eosinophilia are additional manifestations. Poor nutrition may compromise cellular immunity and has been considered an important predisposing condition to developing PCM, especially in younger adults with extensive abdominal lymphadenopathy. (Mycopathologia. 2006 Feb;161(2):73-81) As is typical in acute cases, in our case no pulmonary compromise was observed, but there was mucosal compromise in the colon (figure D), associated with bloody diarrhea. Our patient presented anemia related to the disease itself but also aggravated by gastrointestinal losses due to colonic mucosa compromise. The finding of an O2 Sat of 94% is considered normal when at high altitude, Cusco is at 3200 mt above sea level. A cardinal presentation in our patient was massive splenomegaly on presentation. The differential diagnosis of severe hypersplenism (massive splenomegaly) would include visceral leishmaniasis (not present in Perú, but present in Brazil, Venezuela and Colombia), schistosomiasis (not present in Perú, but present in Brazil, Surinam and Venezuela), chronic brucellosis, hematologic malignancies, and hemophagocytic syndromes. Hyper-reactive malarial splenomegaly syndrome (HMSS) represents one of the leading causes of massive splenomegaly in malaria-endemic countries (Lancet. 2002 Aug 10;360(9331):449-54). Other causes of severe and potentially massive splenomegaly include EBV infection, infective endocarditis, and mycobacterial infections. Disseminated histoplasmosis does not usually cause as massive splenomegaly. Sulfonamides, ketoconazole, itraconazole, and amphotericin B are all effective therapies. Amphotericin B induction therapy for 2 – 4 weeks should be reserved for severe cases or relapses such as this one prior to initiation of itraconazole. For milder cases, itraconazole 100-200 mg/day is the treatment of choice without Amphotericin B induction. A total length of therapy of no less than one year is regarded as the treatment of choice for both the juvenile and chronic forms when it is available and affordable. In resource constraint settings ketoconazole and TMP/SMX become attractive alternatives at a fraction of the cost although in those cases expert opinion will favor at least 12 months of therapy. In this severe and relapsing case, the plan is to give amphotericin B for one month, follow by at least one-year with itraconazole or TMP/SMX. In some instances, lifelong suppressive treatment is a consideration. |